Ehlers-Danlos-Syndrom äußert sich durch überstreckbare Gelenke, Blutergüsse und fragile Haut
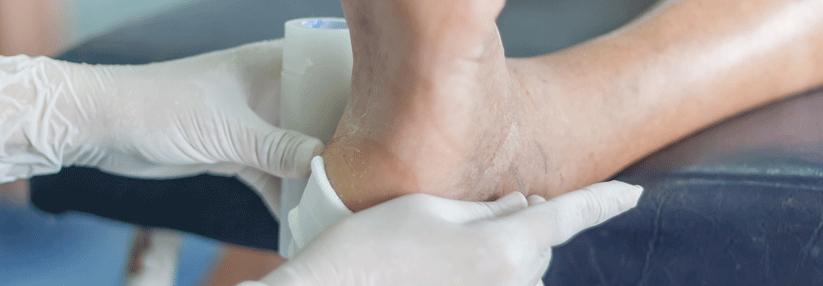
 Typisch für das Ehlers-Danlos-Syndrom sind überstreckbare Gelenke und eine überdehnbare Haut.
© wikimedia/Piotr Dołżonek (CC BY-SA 4.0), Dagger9977 (CC BY-SA 3.0)
Typisch für das Ehlers-Danlos-Syndrom sind überstreckbare Gelenke und eine überdehnbare Haut.
© wikimedia/Piotr Dołżonek (CC BY-SA 4.0), Dagger9977 (CC BY-SA 3.0)
Das Ehlers-Danlos-Syndrom (EDS) ist eine heterogene Gruppe von Erbkrankheiten. Bei 12 von 13 Typen ist die genetische Grundlage bekannt, es kommt aufgrund einer Genmutation zu einer Störung verschiedener Synthesewege, die das Kollagen, die Myomatrix oder das Glykosaminoglykan betreffen.
Patienten mit EDS fallen oft durch eine überdehnbare Haut und Gelenküberstreckbarkeit auf. Da Letztere aber bei 10–30 % der Menschen auftritt, ist sie als alleiniges Kriterium nicht sinnvoll. Je nach EDS-Typ und Patient können auch Gefäße, Muskeln, Bänder, Sehnen sowie innere Organe betroffen sein. Im Praxisalltag haben viele Ärzte die Erkrankung aufgrund der Seltenheit nicht auf dem Schirm, schreiben Dr. Neeti Ghali vom North West Thames Regional Genetics Service and National EDS Service in London und Kollegen und beschäftigten sich ausgiebig mit der 2017 erneuerten internationalen Klassifikation, die weitaus umfassender und spezifischer ist als ihre beiden Vorgänger.
Die drei häufigsten Ehlers-Danlos-Typen
Der Klassiker
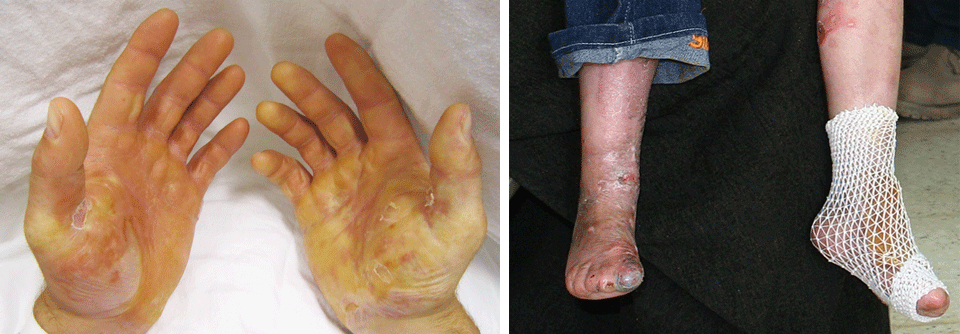
Typisch ist eine fragile und leicht verletzliche Haut, die sich an Knien, Ellbogen und Nacken bis zu 3 cm überdehnen lässt. Hinzu kommen atrophe Narben, Neigung zu Blutergüssen sowie eine generalisierte Gelenkhypermobilität. Das klassische EDS macht zusammen mit dem hypermobilen Typ 90 % der Erkrankungsfälle aus. Der Hypermobile
Einziger Typ, für den es bisher keinen Gentest gibt. Er zeichnet sich vor allem durch eine Überstreckbarkeit der großen und kleinen Gelenke aus, eine Hautfragilität ist eher unüblich. Dagegen sind Subluxationen und Luxationen häufig und meist besteht eine Gelenkinstabilität. Die Betroffenen leiden oft unter (chronischen) Schmerzen. Der Vaskuläre
Äußerlich fällt eine dünne, transparente Haut mit prominenten Venen und einer ausgeprägten Hämatom-Neigung auf, die an Händen und Füßen stark gealtert wirkt. Gesichtsmerkmale sind eine schmale Nase, große Augen und fehlende Ohrläppchen. Ebenfalls typisch sind distale Hypermobilität und Klumpfuß. Etwa 8 von 10 Patienten hatten bis zum 40. Lebensjahr bereits eine Komplikation wie Dissektionen oder Rupturen von Gefäßen und Organen (Darm, Milz, Uterus) oder Pneumothorax.
Typisch ist eine fragile und leicht verletzliche Haut, die sich an Knien, Ellbogen und Nacken bis zu 3 cm überdehnen lässt. Hinzu kommen atrophe Narben, Neigung zu Blutergüssen sowie eine generalisierte Gelenkhypermobilität. Das klassische EDS macht zusammen mit dem hypermobilen Typ 90 % der Erkrankungsfälle aus. Der Hypermobile
Einziger Typ, für den es bisher keinen Gentest gibt. Er zeichnet sich vor allem durch eine Überstreckbarkeit der großen und kleinen Gelenke aus, eine Hautfragilität ist eher unüblich. Dagegen sind Subluxationen und Luxationen häufig und meist besteht eine Gelenkinstabilität. Die Betroffenen leiden oft unter (chronischen) Schmerzen. Der Vaskuläre
Äußerlich fällt eine dünne, transparente Haut mit prominenten Venen und einer ausgeprägten Hämatom-Neigung auf, die an Händen und Füßen stark gealtert wirkt. Gesichtsmerkmale sind eine schmale Nase, große Augen und fehlende Ohrläppchen. Ebenfalls typisch sind distale Hypermobilität und Klumpfuß. Etwa 8 von 10 Patienten hatten bis zum 40. Lebensjahr bereits eine Komplikation wie Dissektionen oder Rupturen von Gefäßen und Organen (Darm, Milz, Uterus) oder Pneumothorax.
Manche Patienten haben blaue Skleren
Die erste Anlaufstelle für viele EDS-Patienten ist und bleibt der Hausarzt. Bei der Anamnese sollten Sie deshalb, neben dem familiären Auftreten eines EDS, gezielt nach folgenden Beschwerden fragen:- Gelenkprobleme (z.B. Schmerzen, Dislokationen, Subluxationen, Hypermobilität)
- Hautprobleme, wie häufige Blutergüsse, schlechte Wundheilung, (atrophe) Narbenbildung
- Hernien, Pneumothorax, Brustwand- oder Wirbelsäulendeformation
- Gefäßrupturen/Organschäden, Aneurysma oder Dissektion in der Vorgeschichte oder bei einem Angehörigen ersten Grades
- Müdigkeit/Erschöpfung
- gastrointestinale Beschwerden (z.B. Hiatushernie, Prolaps, veränderte Motilität)
- posturales Tachykardiesyndrom (POTS) oder andere autonome Dysfunktionen
Das sollten Sie ausschließen
- erbliche Gewebestörungen: Marfan-Syndrom, Loeys-Dietz-Syndrom, familiäres thorakales Aortenaneurysma
- rheumatoide Arthritis
- Misshandlungen, Gerinnungsstörungen als Ursache der Hämatome
- neuromuskuläre Störungen, z.B Kollagen-VI- und -XII-assoziierte Muskeldystrophien
- Bindegewebsstörungen als Ursache für Hypermobilität bzw. Hautelastizität, z.B. Skelettdysplasien (Glasknochen, Achondroplasie) bzw. Pseudoxanthoma elasticum
- andere genetische Ursachen (Mukopolysaccharidose, Noonan-Syndrom)
Man kann die Erkrankung bis dato zwar nicht heilen, aber symptomatisch therapieren:
- Überwachung des kardiovaskulären Systems (insbesondere bei vaskulärem Typ)
- Physio- und Ergotherapie
- orthopädische Hilfsmittel (z.B. Orthesen, Bandagen)
- Tragen von eng anliegenden Langarmshirts oder Leggings unter der Kleidung als Schutz vor Verletzungen der Haut (vor allem bei Kindern)
- spezielle Wundversorgung durch plastische Chirurgen
- Schmerzmedikation (insbesondere bei hypermobiler Form)
Quelle: Ghali N et al. BMJ 2019; 366: I4966; DOI: doi.org/10.1136/bmj.l4966