S1-Leitlinie wird überarbeitet
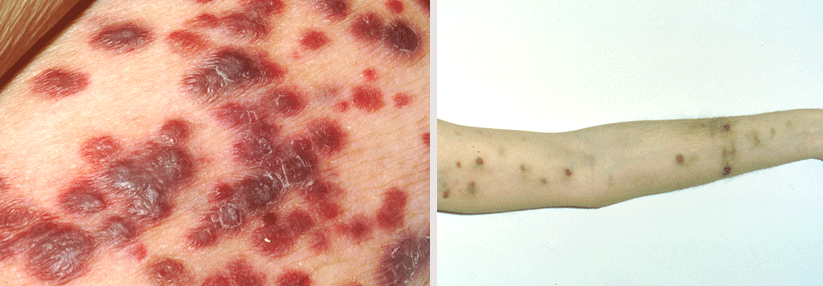
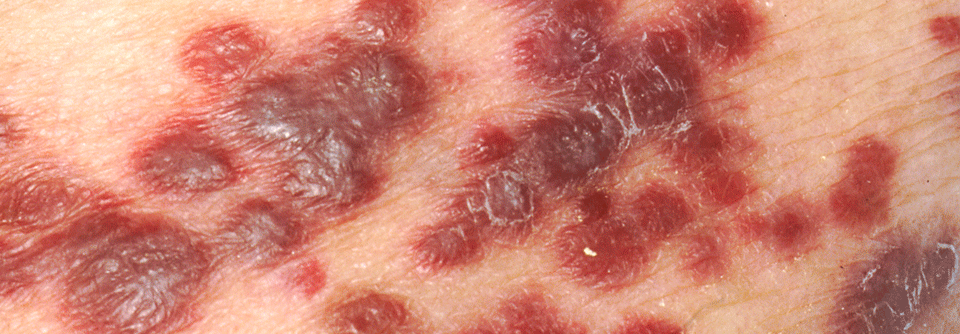 Das klassische Kaposi-Sarkom wird aufgrund seiner Verbreitung auch oft „europäisches“ Kaposi-Sarkom genannt.
© National Cancer Institute, AV-8500-3620
Das klassische Kaposi-Sarkom wird aufgrund seiner Verbreitung auch oft „europäisches“ Kaposi-Sarkom genannt.
© National Cancer Institute, AV-8500-3620
Aktuell wird die S1-Leitlinie Kaposi-Sarkom überarbeitet. Künftig soll man zwischen fünf Subtypen differenzieren. Sie unterscheiden sich durch eine variable Klinik mit akut fudroyanten und aggressiven bis hin zu jahrelang stabilen Verläufen und damit auch prognostisch.
Das klassische sporadische Kaposi-Sarkom tritt vor allem bei älteren Männern in Regionen mit hoher HHV-8-Prävalenz wie Osteuropa oder dem Mittelmeerraum auf, informierte Privdozent Dr. Stefan Esser, Klinik für Dermatologie und Venerologie, Universitätsklinikum Essen. Es befällt insbesondere die unteren Extremitäten, seltener Schleimhäute und innere Organe. Der Verlauf ist indolent, sodass die meisten nicht daran sterben.
Ein Kaposi-Sarkom unter iatrogener Immunsuppression ist in erster Linie durch einen Hautbefall charakterisiert. Nach Reduktion bzw. Absetzen der Immunsuppression ist durchaus eine Regression des Befalls oder sogar eine Heilung möglich.
Das afrikanische (endemische) Kaposi-Sarkom findet sich in der Subsahara-Region überwiegend bei HIV-negativen Menschen. Bei (Klein-)Kindern nimmt es einen aggressiven, oft sogar fulminanten Verlauf mit Lymphadenopathie und Beteiligung der viszeralen Organe ohne Hautbefall. Die Prognose der Betroffenen ist sehr ungünstig.
Dagegen ähnelt der Verlauf bei Erwachsenen, vor allem jungen Männern, i.d.R. dem des klassischen Kaposi-Sarkom. Es kann jedoch auch zu einem aggressiv lokalisierten Verlauf mit Infiltration in Weichgewebe und Knochen kommen. In diesen Fällen ist der Ausgang oft innerhalb von fünf bis sieben Jahren fatal.
Das HIV-assoziierte (epidemische) Kaposi-Sarkom tritt bei HIV-infizierten Personen als Erkrankung mit multiplen Läsionen in Gesicht, Körperstamm und Extremitäten auf. In etwa 20 % der Fälle sind auch die Schleimhäute, in rund 15 % innere Organe betroffen. Zudem besteht die Tendenz zu tumorassoziierten Ödemen.
Die Schwere des Verlaufs hängt von der Zahl CD4-positiver Zellen ab. In der Ära vor der standardmäßigen antiretroviralen Therapie (ART) war der Verlauf aggressiver, ein viszeraler Befall häufig. Doch bereits die Einleitung einer ART führt zu einer deutlichen Besserung, oft auch zur Abheilung, sodass die Erkrankung meist zunächst ohne Ergreifen weiterer Interventionen nur beobachtet wird.
Eine Besonderheit hierbei ist der Immunrekonstitutions-Inflammations-Syndrom(IRIS)-assoziierte Subtyp, bei dem sich ein vorhandenes Kaposi-Sarkom nach Einleitung einer ART bei bisher unbehandelten HIV-positiven Patienten verschlechtern oder subklinische Verläufe manifest werden. Das Risiko für das Auftreten oder eine Aggravierung des Kaposi-Sarkom im Rahmen eines IRIS liegt bei 6,4 %. Besonders hoch fällt das Risiko bei niedriger CD4-Zellzahl von weniger als 100 Zellen/µl vor ART-Beginn aus. Bei HIV-Infizierten mit hohem IRIS-Risiko wird daher teilweise begleitend zur ART eine Chemotherapie initiiert, um die Exazerbation des Kaposi-Sarkom zu verhindern. Beim demaskierenden IRIS-Kaposi-Sarkom ist unverzüglich eine Chemotherapie indiziert.
Als neuer fünfter Typ wurde in die Leitlinie ein Kaposi-Sarkom bei homosexuell aktiven Männern ohne HIV-Infektion aufgenommen. Die Betroffenen sind jünger als die mit klassischem Kaposi-Sarkom und nicht immundefizient. Der Verlauf ist indolent und überwiegend ohne disseminierten Befall und entspricht damit weitgehend dem des klassischen Typs.
Zugrunde liegende Pathophysiologie
Das Kaposi-Sarkom ist ein Tumor lymphatischer Endothelzellen und kann neben Haut und Schleimhäuten auch innere Organe befallen. Es entsteht im Rahmen einer Mehrschritt-Onkogenese, die durch abnormale Neoangiogenese, Inflammation und Proliferation lymphoendothelialer Tumorzellen charakterisiert ist. Als ursächlich für die Entwicklung eines KS gelten in über 95 % der Fälle Infektionen mit dem humanen Herpesvirus 8, das die maligne Transformation auslöst. Immunsuppression und Immundefizienz begünstigen zudem die Entwicklung eines Kaposi-Sarkoms.
Eine universell akzeptierte Stadieneinteilung der fünf Subtypen gibt es laut Dr. Esser bislang nicht. Experten der europäischen Leitlinie empfehlen, beim Management zwischen lokalem, nicht aggressivem Kaposi-Sarkom, lokal aggressiven Formen und disseminiertem Verlauf zu differenzieren.1 Dr. Esser kritisierte, dass hierbei weder Immunstatus noch Alter, Schleimhaut-, viszeraler und rein kutaner Befall berücksichtigt werden. Laut deutscher Leitlinie sollte künftig das Management individualisiert in Abhängigkeit der aufgeführten Prognosefaktoren erfolgen.
Für die histologische Sicherung eines klinischen Typs ist spätestens vor Einleitung einer spezifischen Therapie eine tiefe Biopsie erforderlich. Patienten ohne bekannte HIV-Infektion muss bei der Erstdiagnose ein HIV-Test angeboten werden. Als unverzichtbar bezeichnete Dr. Esser weiterhin die Ausbreitungsdiagnostik mit vollständiger Inspektion der einsehbaren Schleimhäute, Palpation der Lymphknoten und des Abdomens sowie Erfassung aller Läsionen und Symptome. Die weiteren Untersuchungen werden anschließend individualisiert je nach Befall, Symptomatik und Subtyp durchgeführt. Bei Verdacht auf einen viszeralen Befall spricht sich die überarbeitete S1-Leitlinie für eine Ganzkörper-CT aus.
Als Ziel der Behandlung nannte der Referent die Rückbildung der Läsionen, die Kontrolle über den Krankheitsverlauf und die Besserung der Symptome bei Erhalt sowohl der Lebensqualität als auch der Lebenserwartung. Allerdings gibt es bisher kein allgemein anerkanntes Standard-Therapieschema. Im Fall einer guten Prognose und engmaschigen Nachbeobachtung ist eine sofortige spezifische Therapie nicht in allen Fällen erforderlich. Einzelne störende Läsionen können lokal, z.B. durch Exzision oder Kryotherapie, intraläsionale Injektion eines Vinca-Alkaloids oder topisch behandelt werden.
Aufgrund der hohen Strahlensensitivität des Kaposi-Sarkoms eignet sich zudem eine Radiatio. Bei Patienten mit Lymphödemen empfahl Dr. Esser begleitend Lymphdrainage und Kompressionstherapie. Im Rahmen der systemischen Therapie gilt pegyliertes Doxorubicin als Mittel der Wahl. Alternativ kann auch pegyliertes Daunorubicin verwendet werden. Paclitaxel sollte bei Versagen von pegyliertem Doxorubicin in der zweiten Linie zum Einsatz kommen.
1. Lebbe C et al. Eur J Cancer 2019; 114: 117-127; DOI: 10.1016/j.ejca.2018.12.036
Quelle: Esser S. 31. Deutscher Hautkrebskongress
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).
 Das klassische Kaposi-Sarkom wird aufgrund seiner Verbreitung auch oft „europäisches“ Kaposi-Sarkom genannt.
© National Cancer Institute, AV-8500-3620
Das klassische Kaposi-Sarkom wird aufgrund seiner Verbreitung auch oft „europäisches“ Kaposi-Sarkom genannt.
© National Cancer Institute, AV-8500-3620