Phäochromozytom Erst entdeckt, dann gen-gecheckt
 Zu einer guten Behandlung des Phäochromozytom gehören auch die Nachsorge und eine genetische Diagnostik.
© hywards – stock.adobe.com
Zu einer guten Behandlung des Phäochromozytom gehören auch die Nachsorge und eine genetische Diagnostik.
© hywards – stock.adobe.com
Charakteristisch für das Phäochromozytom ist die vermehrte Sekretion von Katecholaminen. Unter den zufällig entdeckten adrenalen Tumoren hat es einen Anteil von etwa 5 %. Die adrenalen Tumoren sind zwar potenziell maligne, bleiben aber meist gutartig. In etwa 10 % erfolgt eine bilaterale Manifestation und in etwa 30-40 % liegen pathogene genetische Veränderungen vor. Am bekanntesten sind drei Formen:
- Multiple endokrine Neoplasie Typ 2 (MEN2),
- Von-Hippel-Lindau-Syndrom und
- Neurofibromatose 1.
Die Symptomatik des Phäochromozytoms richtet sich nach dem vermehrt produzierten Katecholamin. Die klassische Triade aus anfallsartigen Kopfschmerzen, Schwitzen und Palpitationen weisen nur etwa 20 % der Patienten auf. Aber fast alle haben mindestens eine dieser Beschwerden, schreiben Prof. Dr. Nicolas Schlegel vom Universitätsklinikum Würzburg und Koautoren. Zum klinischen Bild gehören auch Hypertonie, Tachykardie, Blässe und Angst. Infolge des Hochdrucks können Herzinsuffizienz, Myokardinfarkt, Schlaganfall sowie spontane Blutungen und dissezierende Aneurysmen auftreten.
Die Abklärung erfolgt im ersten Schritt mit der biochemischen Bestimmung der freien Plasmametanephrine oder der fraktionierten Metanephrine im 24-Stunden-Urin. Vor der Blutentnahme sollte etwa eine halbe Stunde liegen. Bei einem Ruhewert über der dreifachen Norm ist der Nachweis erbracht. In grenzwertigen oder unklaren Fällen kann ein Clonidinhemmtest für Klarheit sorgen. Vor der Diagnostik sollten interferierende Medikamente wie Paracetamol, MAO-Hemmer und trizyklische Antidepressiva abgesetzt werden. Weil bis zu 40 % der Phäochromozytome mit genetischen Veränderungen assoziiert sind, sollten entsprechende Tests angeboten werden.
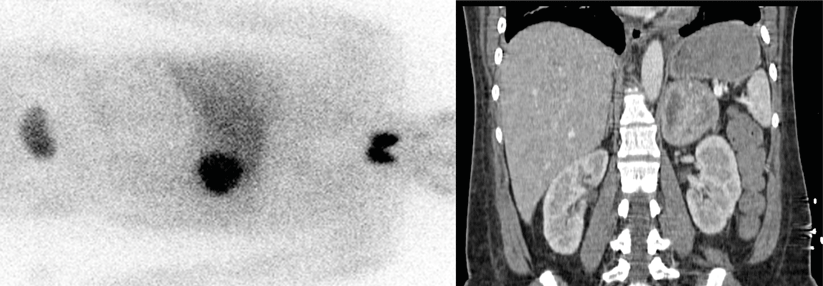
Zur Lokalisation kommen vor allem Schnittbildverfahren wie CT oder MRT zum Einsatz. Eine funktionelle nuklearmedizinische Diagnostik ist laut Leitlinien nicht mehr zwingend erforderlich. Sie ermöglicht aber die Detektion von Metastasen und beeinflusst so das therapeutische Konzept. Eine präoperative PET/CT wird angeraten bei einer Tumorgröße > 5 cm, erhöhten Plasmawerten von 3-Methoxythyramin (3MT) oder bekannter Keimbahnmutation im SDHB*-Gen.
Die präoperative Alphablockade soll eine hämodynamische Instabilität während des Eingriffs vermeiden und perioperativen Komplikationen vorbeugen. Ihr Nutzen wird aber zunehmend kontrovers diskutiert. Prof. Schlegel und Kollegen schätzen, dass die perioperative Blutdruckeinstellung bei der inzwischen erfolgten Reduktion der peripheren Mortalität unter 1 % nur eine untergeordnete Rolle spielt. Viel wichtiger ist die Operation in einem spezialisierten Zentrum. Außerdem sollten individuelle Faktoren wie Tumorgröße und schwer einstellbarer Blutdruck bei der Entscheidung pro und kontra Alphablockade berücksichtigt werden.
Konsens besteht, dass die meisten Nebennierentumoren minimalinvasiv entfernt werden sollten. Wichtig ist, dass sich die Geschwulst ohne Kapselruptur sicher entfernen lässt. Dann ist auch eine Größe über 6–10 cm kein Hindernis. Laparoskopische und retroperitoneoskopische Verfahren sind gleichwertig.
Eine kurative Therapie ist nur operativ möglich. Bei bilateralen Phäochromozytomen bietet eine parenchymerhaltende Resektion den Vorteil, dass die Nebennierenrinde funktionstüchtig bleiben kann. Dazu muss mindestens ein Drittel des hormonsezierenden Gewebes belassen werden. In einer Registerstudie gelang dies in mehr als 70 % der Fälle. Allerdings müssen die Vorteile dieser Strategie prinzipiell gegen die Rezidivgefahr abgewogen werden. Metastasierungsrisiko und Gesamtsterblichkeit sind unter vollständiger und partieller Adrenalektomie im Allgemeinen vergleichbar. Vorsicht ist bei Patienten mit SDH-B-Mutation geboten: Bei diesen übersteigt das Malignitätsrisiko nach organerhaltendem Eingriff teilweise 50 %.
Elementar für die Prognose ist eine gute Nachsorge. Patienten ohne genetische Belastung sollten sich zehn Jahre lang einmal jährlich untersuchen lassen. Bei Mutationen oder anderen Risikofaktoren (z.B. großer Tumor, sehr junges Alter) ist eine lebenslange Überwachung indiziert. Die erste Kontrolle wird zwei bis sechs Wochen nach dem Eingriff empfohlen. Sie dient dem Nachweis, dass sich die präoperativ erhöhten Metanephrine normalisiert haben, was die vollständige Resektion bestätigt.
Spätesten jetzt sollte allen Patienten eine genetische Diagnostik angeboten werden, denn diese beeinflusst das Management. Je nach Veränderung fokussiert sich die Nachsorge auf unterschiedliche Erkrankungen. Bei der RET-Mutation sind das primärer Hypoparathyreoidismus und medulläres Schilddrüsenkarzinom. Im Fall einer SDHx-Mutation entwickeln sich vermehrt hormoninaktive Paragangliome (auch im Kopfhalsbereich). Für eine optimale Patientenversorgung raten die Autoren, alle Patienten mit Phäochromozytom in einem interdisziplinären endokrinen Tumorboard vorzustellen und für eine sichere Anbindung an den nachbehandelnden Endokrinologen zu sorgen.
* Succinate Dehydrogenase Complex Iron Sulfur Subunit B
Quelle: Schlegel N et al. Chirurgie 2024; 95: 200-206; DOI: 10.1007/s00104-023-01988-6