Lupus oder Lymphom? Wann der Hämatologe mit ins Boot muss
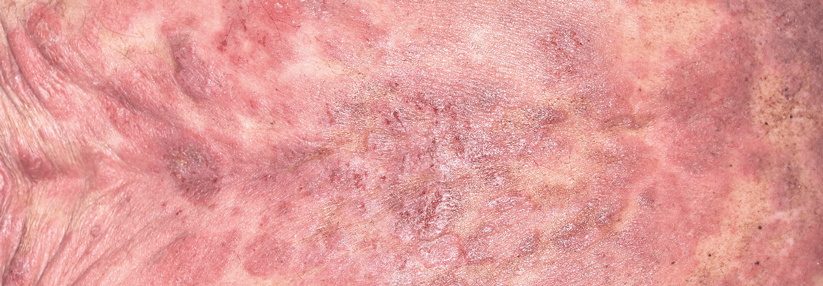
 In seltenen Fällen kann hinter einer Pannikulitis auch ein kutanes T-Zell-Lymphom stecken.
© SciePro – stock.adobe.com
In seltenen Fällen kann hinter einer Pannikulitis auch ein kutanes T-Zell-Lymphom stecken.
© SciePro – stock.adobe.com
Spricht eine Pannikulitis, ein systemischer Lupus erythematodes (SLE) oder eine andere Bindegewebserkrankung (connective tissue disease, CTD) nicht wie erwartet auf die medikamentöse Therapie an, sollte man die ursprüngliche Diagnose kritisch hinterfragen. In seltenen Fällen liegt statt der vermuteten rheumatologischen Erkrankung eine hämatologische vor: ein kutanes T-Zell-Lymphom (CTCL). Denn hinsichtlich der systemischen Manifestationen von Autoimmun- und lymphoproliferativen Erkrankungen gibt es Überschneidungen, erklärte Dr. Tali Eviatar vom Tel Aviv Sourasky Medical Center. Zusammen mit ihrer Kollegin Dr. Katya Meridor präsentierte sie zwei Fälle aus ihrer Klinik, die man fälschlicherweise für CTD-Fälle gehalten hatte. Bei beiden Patienten wurde im Verlauf die Diagnose korrigiert: Sie litten nicht, wie ursprünglich gedacht, an einem Morbus Sjögren oder einem SLE, sondern an einem primären kutanen gamma/delta-positiven T-Zell-Lymphom. Die Patienten starben, trotz mehrerer Therapieversuche, einige Monate nach der (korrekten) Diagnose.
Systemische Lymphome wie die chronische lymphatische Leukämie, B-Zell-Lymphome, T-Zell-Lymphome und das multiple Myelom führen mitunter zu Beschwerden, die einer CTD stark ähneln, so Dr. Eviatar. Neben der klassischen B-Symptomatik (Fieber, Gewichtsverlust, Nachtschweiß) kann es zu einer autoimmunhämolytischen Anämie, einer Immunthrombozytopenie und anderen Zytopenien kommen. Typische Hauterscheinungen sind das Sweet-Syndrom, die Pannikulitis und die Vaskulitis. Daneben können Kryoglobulinämie, Amyloidose und Lymphadenopathie auftreten. Gerät die Inflammation außer Kontrolle, kommt es zur hämophagozytischen Lymphohistiozytose (HLH) oder einem Makrophagenaktivierungssyndrom (MAS).
Bei Patienten mit einer lobulären lymphozytären Pannikulitis gibt es laut der Referentin drei wichtige Differenzialdiagnosen: die Lupus-Pannikulitis, das subkutane pannikulitisartige T-Zell-Lymphom (SPTCL) und das kutane gamma/delta-positive T-Zell-Lymphom. Den Unterschied zu erkennen, ist nicht trivial. Bei der Lupus-Pannikulitis sind meist eher der Körperstamm und die oberen Extremitäten sowie das Gesicht betroffen, so die Kollegin. Ein Befall der Kopfhaut und der unteren Extremitäten spricht eher für ein Lymphom. Allerdings sind in beiden Fällen Ausnahmen möglich. Zu einer HLH kommt es beim SPTCL oder dem kutanen gamma/delta-positiven T-Zell-Lymphom in bis zu 50 % der Fälle. Beim Lupus ist das Hyperinflammationssyndrom eher selten. Auch histopathologisch und immunhistochemisch gibt es Unterschiede. Eine enge Zusammenarbeit mit Pathologen, Hämatologen und Dermatologen ist unabdingbar für die korrekte Diagnose und eine bestmögliche Betreuung der Patienten, so Dr. Eviatar.
Quelle: EULAR 2023 – Annual European Congress of Rheumatology