Auch eine leichte Hämolyse sollte den Verdacht auf Kugelzellanämie wecken
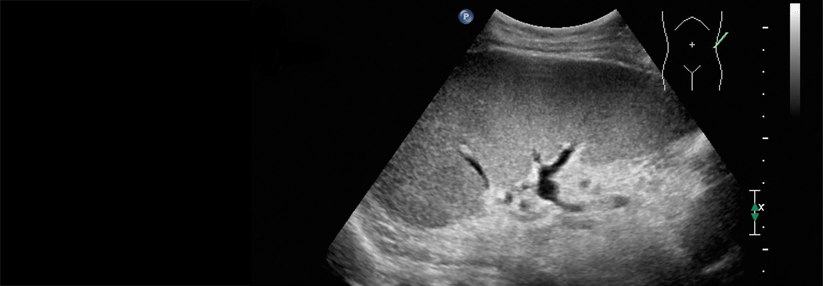 Ein Gendefekt wird oft nur zufällig entdeckt.
© Albertinen-Krankenhaus Hamburg/sonographiebilder.de
Ein Gendefekt wird oft nur zufällig entdeckt.
© Albertinen-Krankenhaus Hamburg/sonographiebilder.de
Ursache der angeborenen Kugelzellanämie sind Mutationen unterschiedlicher Strukturgene. Diese führen dazu, dass bestimmte, zur Stabilität der Erythrozytenmembran notwendige Proteine fehlerhaft gebildet werden. Daraus resultiert eine verminderte Verformbarkeit der Erythrozyten und ein beschleunigter lienaler Abbau, erklären Professor Dr. Stefan W. Eber von der Kinderklinik der Technischen Universität München und Dr. Oliver Andres von der Universitäts-Kinderklinik Würzburg in der aktualisierten S1-Leitlinie „Hereditäre Sphärozytose“.
Je nach Lebensalter und genetischer Veränderung variiert die Symptomatik der hereditären Sphärozytose (HS) stark. So gibt es klinisch gesunde Erwachsene mit einer mild ausgeprägten kompensierten Hämolyse ohne Anämie, aber auch Säuglinge und Kleinkinder, die aufgrund einer schweren Verlaufsform regelmäßig Transfusionen benötigen. Beim autosomal-rezessiven Erbgang werden manche Anlageträger mangels klinischer Symptome erst dann identifiziert, wenn aus anderen Gründen eine Blutentnahme erfolgt oder die Familienanamnese positiv ist. In den meisten Fällen ist er aber mit einem schweren klinischen Phänotyp assoziiert. Folgende Befunde gelten als Leitsymptome der Kugelzellanämie:
Eine symptomatische Therapie der Kugelzellanämie ist meist nicht notwendig, mit Ausnahme von Erythrozytentransfusionen in den beiden ersten Lebensjahren und bei aplastischen Krisen. Eine kausale Behandlung wäre zwar durch eine allogene Stammzelltransplantation vorstellbar, doch diese hat derzeit keinen Stellenwert.
Häufigste angeborene hämolytische Anämie
Bei rund zwei Dritteln der Betroffenen liegt eine gesicherte familiäre autosomal-dominante Form vor. Bei etwa jedem Vierten ist es zu Neumutationen in der mütterlichen Keimbahn gekommen (meist autosomal-dominante Defekte). Ein autosomal-rezessiver Erbgang findet sich in 10 % der Fälle.Schweregrade der hereditären Sphärozytose (HS) | ||||
|---|---|---|---|---|
| Leichte HS | Mittelschwere HS | Schwere HS1 | Sehr schwere HS2 | |
| Anteil an Patienten (%) | 25−33 | 60−70 | ca. 10 | 3−4 |
| Hämoglobin (g/dl) | 11,0−15,0 | 8,0−11,0 | 6,0−9,0 | < 6,0 |
| Retikulozyten (%) | 1,5−6 | ≥ 6 | ≥ 10 (meist > 15)3 | ≥ 10 |
| Bilirubin (mg/dl) | 1−2 | ≥ 2 | 2−3 | ≥ 3 |
| Sphärozyten (Blutausstrich) | oft nur vereinzelt | deutlich vermehrt | deutlich vermehrt | Mikrosphärozyten undPoikilozyten |
1Patienten benötigen in den ersten beiden Lebensjahren gehäuft bzw. regelmäßige Transfusionen. 2Patienten müssen regelmäßige Transfusionen bekommen, um einen Hämoglobinwert über 6,0 g/dl zu halten. 3Die Retikulozytenzahl ist infolge der nach der Trimenonreduktion verzögert einsetzenden Erythropoese z.T. nur mäßig erhöht. nach Eber S, Andres O, S1-Leitlinie Hereditäre Sphärozytose; AWMF-Register Nr. 025/018 | ||||
Je nach Lebensalter und genetischer Veränderung variiert die Symptomatik der hereditären Sphärozytose (HS) stark. So gibt es klinisch gesunde Erwachsene mit einer mild ausgeprägten kompensierten Hämolyse ohne Anämie, aber auch Säuglinge und Kleinkinder, die aufgrund einer schweren Verlaufsform regelmäßig Transfusionen benötigen. Beim autosomal-rezessiven Erbgang werden manche Anlageträger mangels klinischer Symptome erst dann identifiziert, wenn aus anderen Gründen eine Blutentnahme erfolgt oder die Familienanamnese positiv ist. In den meisten Fällen ist er aber mit einem schweren klinischen Phänotyp assoziiert. Folgende Befunde gelten als Leitsymptome der Kugelzellanämie:
- normozytäre Anämie (in ca. 70 %)
- leicht mikrozytäre Anämie (in rund 30 %)
- Skleren- oder generalisierter Ik-terus (hämolytischer Ikterus, Verschlussikterus)
- Splenomegalie
- Gallensteine, die manchmal schon im Kindesalter Beschwerden verursachen.
Eine symptomatische Therapie der Kugelzellanämie ist meist nicht notwendig, mit Ausnahme von Erythrozytentransfusionen in den beiden ersten Lebensjahren und bei aplastischen Krisen. Eine kausale Behandlung wäre zwar durch eine allogene Stammzelltransplantation vorstellbar, doch diese hat derzeit keinen Stellenwert.
Vorsicht Ringelröteln!
Erkranken Kinder mit hereditärer Sphärozytose an Ringelröteln, kann eine aplastische Krise auftreten. Die Infektion mit dem Parvovirus B19 führt zu einer Retikulopenie mit starkem Hämoglobinabfall und erfordert gegebenenfalls eine Bluttransfusion. Eltern von seronegativen Kindern und noch nicht seropositive Jugendliche oder Erwachsene müssen darauf hingewiesen werden, auf Ringelröteln im Umfeld sowie plötzliche Blässe, ausgeprägte Schwäche oder sehr blasse Bindehaut zu achten. All dies kann nämlich auf die Gefahr einer aplastischen Krise hinweisen. Wichtig zu wissen ist auch, dass der typische Ringelröteln-Ausschlag bei Patienten mit chronisch hämolytischer Anämie fast immer fehlt!
Wirksame Therapie ist die Splenektomie
Schwere und bei gegebener Indikation auch mittelschwere Formen können nämlich wirksam mittels Splenektomie behandelt werden. Diese sollte möglichst nicht vor dem 6. und keinesfalls vor dem 3. Lebensjahr erfolgen. Bei leichter HS ist in der Regel keine Splenektomie im Kindes- und Jugendalter erforderlich. Das Entfernen der Milz führt dazu, dass sich Hämoglobin und Retikulozytenzahl normalisieren. Lediglich bei sehr schwerer HS kann eine leicht gesteigerte Hämolyse bestehen bleiben.Besser ein Stückchen Milz dran lassen
Da nach kompletter Splenektomie lebenslang ein erhöhtes Sepsisrisiko besteht und die Antibiotikaresistenz von Pneumokokken zunimmt, empfehlen die Autoren, die Milz nicht vollständig zu entfernen, sondern eine kleine Restmilz von etwa 10 ml zu belassen. Im Hinblick auf die Immunabwehr und das Thromboembolierisiko bringe dies Vorteile.Nach kompletter oder nahezu vollständiger Milzentfernung soll mehrere Jahre lang eine Prophylaxe mit Penicillin oder Amoxicillin durchgeführt werden, um eine foudroyante Postsplenektomie-Infektion zu verhindern. Vor und nach Splenektomie ist auf einen guten Impfschutz der Patienten zu achten (spezifische Empfehlungen unter www.asplenie-net.org). Eine Cholezystektomie kann bei symptomatischen Gallensteinen notwendig werden.
Quelle: Eber S, Andres O, S1-Leitlinie Hereditäre Sphärozytose; AWMF-Register Nr. 025/018
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).
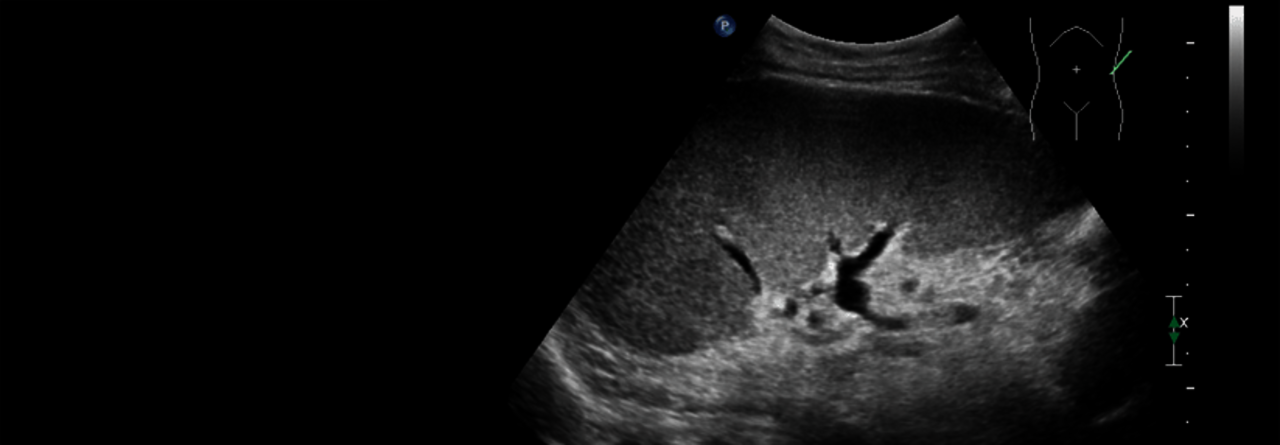 Ein Gendefekt wird oft nur zufällig entdeckt.
© Albertinen-Krankenhaus Hamburg/sonographiebilder.de
Ein Gendefekt wird oft nur zufällig entdeckt.
© Albertinen-Krankenhaus Hamburg/sonographiebilder.de
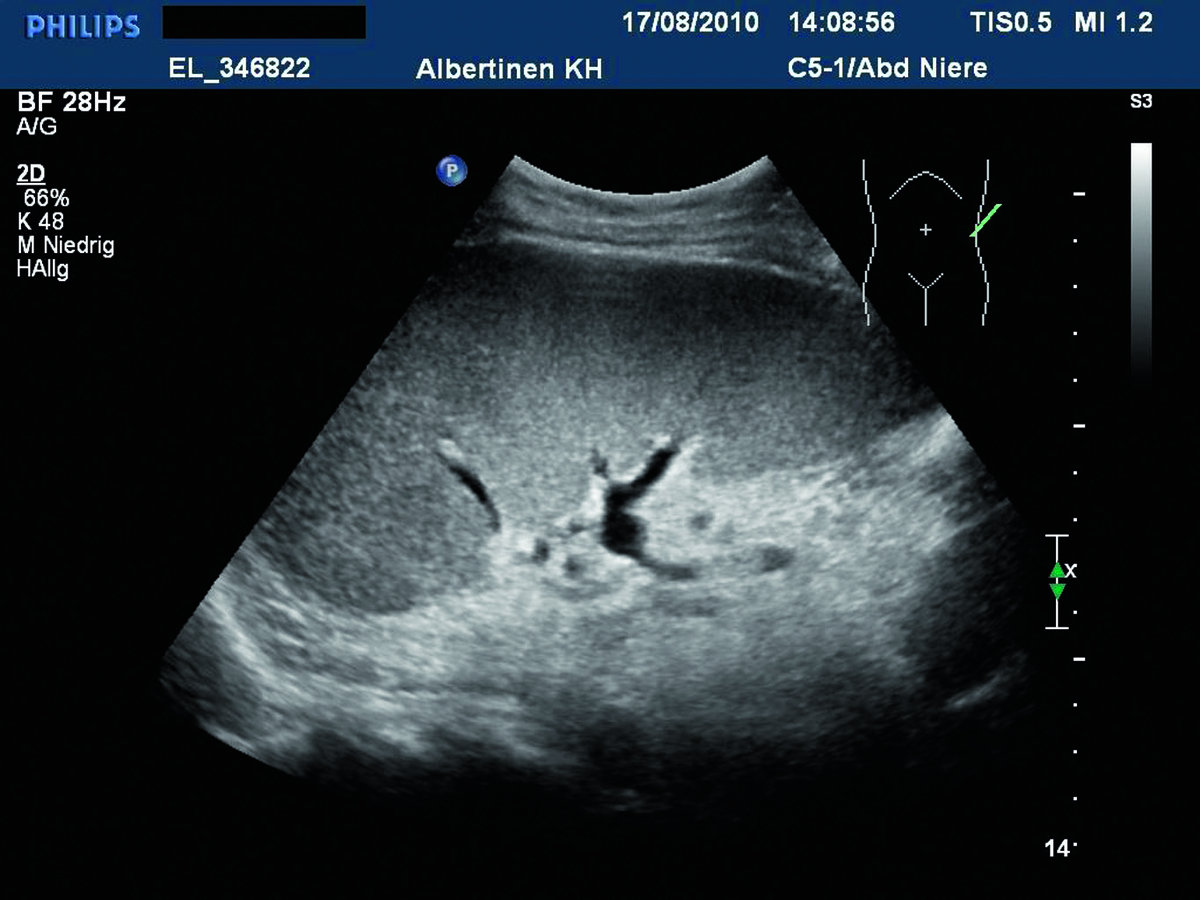 Eines der Leitsymptome der hereditären Sphärozytose ist eine stark vergrößerte Milz.
© Albertinen-Krankenhaus Hamburg/sonographiebilder.de
Eines der Leitsymptome der hereditären Sphärozytose ist eine stark vergrößerte Milz.
© Albertinen-Krankenhaus Hamburg/sonographiebilder.de