Wenn die Lysosomen am Messie-Syndrom leiden
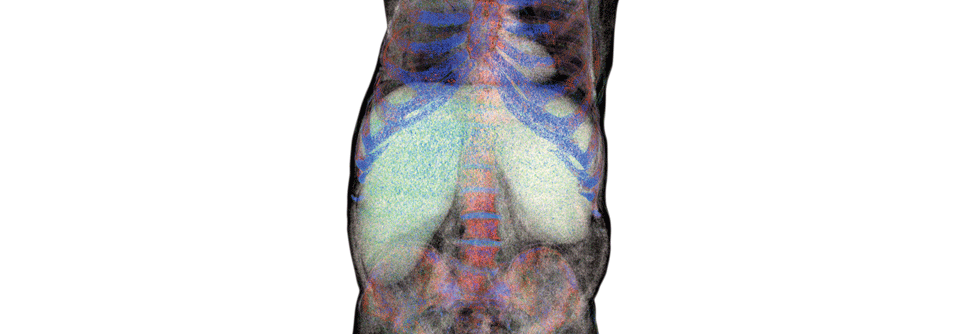 Ein markantes Krankheitszeichen der lysosomalen Speicherkrankheiten ist die Hepatosplenomegalie.
© Science Photo Library/ VSEVOLOD ZVIRYK
Ein markantes Krankheitszeichen der lysosomalen Speicherkrankheiten ist die Hepatosplenomegalie.
© Science Photo Library/ VSEVOLOD ZVIRYK
Als zentrales Stoffwechselorgan ist die Leber bei genetischen Stoffwechselerkrankungen – wie Hämochromatose, Morbus Wilson oder Alpha-1-Antitrypsinmangel – oft betroffen. Doch auch andere Erbkrankheiten wie lysosomale Speicherkrankheiten (LSK) können sich an der Leber manifestieren und eine Hepatosplenomegalie bedingen, schreiben Prof. Dr. Vanessa Stadlbauer-Köllner, Medizinische Universität Graz, und Prof. Dr. Elmar Aigner, Universitätsklinikum Salzburg. Zwar werden die meisten LSK im Kindesalter diagnostiziert, doch können leichtere Verlaufsformen auch erst im Erwachsenenalter auffallen (s. Kasten).
Lysosomale Speicherkrankheiten
Lysosomen sind Zellorganellen, die vom Körper nicht mehr benötigte Makromoleküle verstoffwechseln. Sie enthalten zahlreiche Enzyme, die z.B. Lipide, Mukopolysaccharide etc. abbauen. Funktioniert eines dieser Enzyme nicht mehr richtig oder fehlt es, sammelt sich die abzubauende Substanz an, was verschiedene Störungen nach sich zieht.
Heute sind über 70 Enzymdefekte und damit verschiedene lysosomale Speicherkrankheiten (LSK) bekannt, die jeweils sehr selten sind. Schwere Formen manifestieren sich bereits bei Babys oder Kleinkindern, während sich leichtere Varianten oft erst im Erwachsenenalter bemerkbar machen.
Da man als Mediziner bei Erwachsenen nicht unbedingt eine LSK auf dem Schirm hat und die Symptomatik recht unspezifisch sein kann, verstreicht oft viel Zeit bis zur Diagnosestellung.
Wenn sich also ein Erwachsener mit einer Hepatosplenomegalie in der Praxis vorstellt, sollte man differenzialdiagnostisch auch an folgende Erkrankungen denken:
- Morbus Gaucher
- Mangel an saurer Sphingomyelinase (ASMD), Morbus Niemann-Pick Typ B
- Mangel an lysosomaler saurer Lipase (LAL-D), Cholesterinester-Speichererkrankung
Die Zeichen und Symptome dieser drei seltenen Erkrankungen sind in der Tabelle aufgelistet.
| Symptome der drei lysosomalen Speicherkrankheiten im Überblick | ||
|---|---|---|
| Morbus Gaucher | Mangel an saurer Sphingomyelinase (ASMD) | Mangel an lysosomaler saurer Lipase (LAL-D) |
|
|
|
Spezialisierte Labors bieten Tests mit Trockenblut an
Wie weist man eine LSK nach? Wichtigster Schritt in der Diagnostik ist es, bei Erwachsenen überhaupt an die Möglichkeit einer Speicherkrankheit zu denken, betonen die Kollegen aus Österreich.
Verdacht sollte man schöpfen, wenn Laborbefunde wie Anämie, Thrombopenie, Hyperferritinämie und/oder ein auffälliger Lipidstatus vorliegen, die sich nicht durch eine fortgeschrittene Lebererkrankung oder eine hämatologische Erkrankung mit gesicherter Ätiologie erklären lassen.
Die Verdachtsdiagnose LSK lässt sich über eine Bestimmung der jeweiligen Enzymaktivität in Leukozyten oder Fibroblasten aus einer Hautbiopsie sichern. Es gibt heute aber auch unkomplizierte Testverfahren zur Bestimmung der Enzymaktivität mithilfe von Trockenblut. Sie werden von spezialisierten Labors angeboten.
Mit Multitest gleich auf drei Speicherkrankheiten screenen
Inzwischen steht mit der sogenannten Trockenblutkarte ein Multitest zur Verfügung, der bei unklarer Hepatosplenomegalie mit klinischem Verdacht auf LSK durchgeführt werden sollte. Auf diese Filterkarte wird in markierte Bereiche Blut des Patienten getropft. Anschließend muss sie getrocknet und ans Labor geschickt werden. Mit dem Test kann in einem einzigen Untersuchungsschritt auf M. Gaucher, ASMD und LAL-D gescreent werden. Fällt eine verminderte Enzymaktivität auf, ist eine Sicherung der Diagnose mittels Mutationsnachweis durch Sequenzierung des jeweiligen Gens angezeigt.
Für die LSK stehen wirksame Behandlungen zur Verfügung. Bei der Enzymersatztherapie wird das defekte Enzym durch ein biotechnologisch hergestelltes Enzym ersetzt. In der Regel erfolgt die Behandlung alle zwei Wochen intravenös. Ziel der Substratreduktionstherapie ist es, die Biosynthese des schädlichen oder des nicht mehr abgebauten Metaboliten zu hemmen. Für M. Gaucher ist das in Form einer oralen Therapie möglich.
Die Enzymersatztherapie ist bei M. Gaucher, ASMD und LAL-D recht erfolgreich. Die Lebergröße nimmt ab und die Laborwerte normalisieren oder verbessern sich deutlich. Die Behandlungserfolge bei hepatischen LSK unterstreichen die Bedeutung einer rechtzeitigen Diagnosestellung, fassen die Autoren zusammen.
Quelle: Stadlbauer-Köllner V, Aigner E. internistische praxis 2023; 66: 238-248
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).

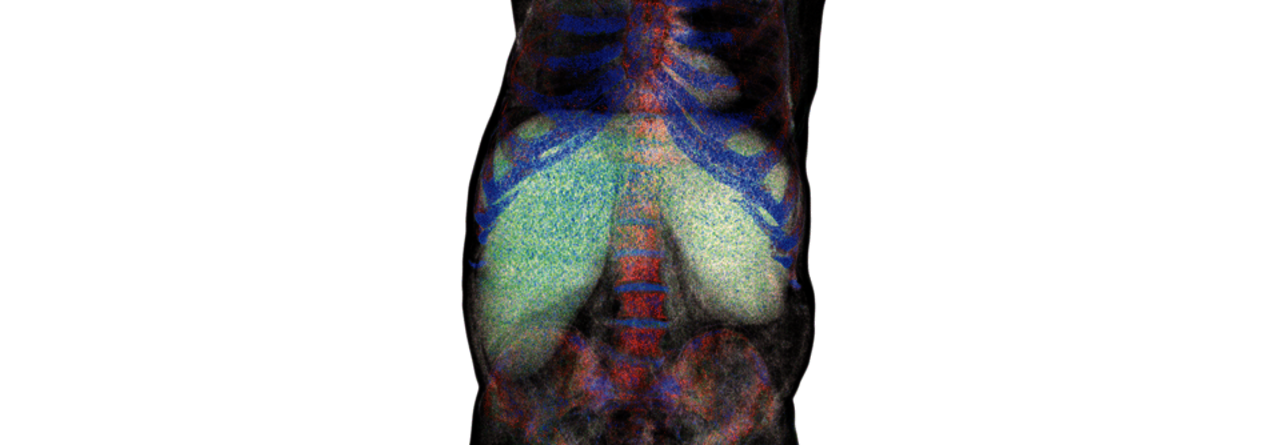 Ein markantes Krankheitszeichen der lysosomalen Speicherkrankheiten ist die Hepatosplenomegalie.
© Science Photo Library/ VSEVOLOD ZVIRYK
Ein markantes Krankheitszeichen der lysosomalen Speicherkrankheiten ist die Hepatosplenomegalie.
© Science Photo Library/ VSEVOLOD ZVIRYK