Genetische Vielfalt von gastroösophagealen Tumoren erschwert individuelle Therapie
 Nur wenn man alle relevanten Mutationen eines Tumors kennt, kann eine individuelle Krebstherapie diese adressieren.
© iStock/teekid
Nur wenn man alle relevanten Mutationen eines Tumors kennt, kann eine individuelle Krebstherapie diese adressieren.
© iStock/teekid
Jede gefundene Treibermutation, zu der es zielgerichtete Medikamente gibt, führt idealerweise zu einer entsprechenden Therapie – je nach Zulassung neoadjuvant, adjuvant und/oder palliativ. Beim gastroösophagealen Adenokarzinom (GEA) beeinflussen beispielsweise Variationen im HER2-, MET- und FGFR2-Gen die Behandlung. Für eine ganze Reihe weiterer Mutationen gibt es bislang aber noch keine zugelassenen Medikamente.
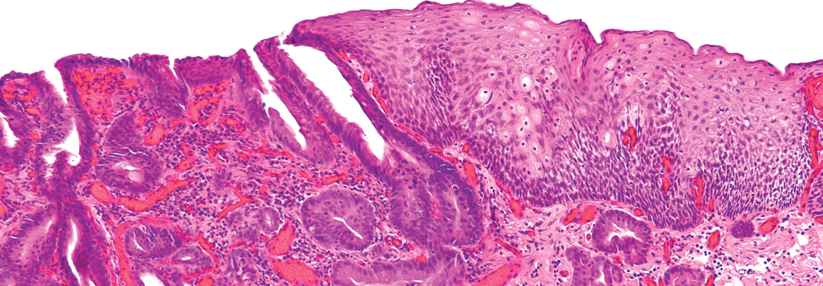
Die räumlich sehr heterogene Verteilung der Mutationen innerhalb dieses Tumors, wie sie etwa im The Cancer Genome Atlas (TCGA) zutage kam, hat sich als Problem erwiesen. Insbesondere im Hinblick auf eine neoadjuvante Therapie, bei der lediglich ein kleines Biopsat verfügbar ist. Mutationen, die nur in Arealen außerhalb der Gewebeprobe vorliegen, werden nicht detektiert und daher auch nicht therapeutisch berücksichtigt. In der Folge wachsen die betroffenen Krebsregionen weiter.
Biopsate auf 891 relevante Onkogene geprüft
Um das Problem analytisch möglichst umfassend anzugehen, requirierten Onkologen, Pathologen und Biostatistiker um Professor Dr. Joseph Chao aus einer Gewebebank des City of Hope Comprehensive Cancer Center in Duarte Operationsmaterial von 41 Patienten, deren GEA zwischen 1989 und 2013 in kurativer Absicht reseziert worden war. Die Daten von 37 Personen flossen in die Analyse ein. Einzelnukleotid- und Kopienzahlveränderungen bestimmten die Wissenschaftler mithilfe eines Arrays, der 891 onkologisch relevante Gene umfasst. Die räumliche Heterogenität erschlossen sie unter anderem mithilfe von Fluoreszenz-in-situ-Hybridisierung (FISH). Die klonale Heterogenität der Tumoren ermittelten die Forscher mittels komplexer bioinformatorischer Programme und definierten einen klonalen Kompositions-Score, der zwischen 0 und 3 liegen konnte.
46 % der Proben wiesen eine hohe intratumorale Heterogenität auf. Insgesamt war ein klonaler Kompositions-Score von mindestens 2 gegenüber einem Score von 0 oder 1 mit einem vierfach erhöhten Mortalitätsrisikos assoziiert (Hazard Ratio [HR] 3,92; 95%-KI 1,27–12,08; p = 0,02).
Fazit
Eine erhebliche räumliche Heterogenität bezüglich der Expression onkogener Alterationen existiert im Primärtumor bereits, bevor eine metastatische Absiedelung stattgefunden hat. Je höher sie ist, desto schlechter fällt die Prognose des resezierten GEA aus. Das unterstreicht die Probleme, die sich im Hinblick auf die Entwicklung zielgerichteter Therapien stellen: Durch mehrere Biopsien kann die Heterogenität zu einem gewissen Grad zwar abgebildet werden, schreiben die Autoren. Bewegt sie sich jedoch im Mikrometerbereich, sind sehr aufwendige Einzelzellanalysen erforderlich, um entsprechende Strategien für Kombinations- und/oder sequenzielle Behandlungen zu entwickeln. Das gilt laut den Wissenschaftlern für zielgerichtete wie auch für Immuntherapien. Die Herausforderung dürfte sich nicht auf gastroösophageale Tumoren beschränken.
Alterationen nur Mikrometer voneinander entfernt
Das Risiko fiel in einer multivariaten Analyse, die Variablen wie Tumorstadium, histologischer Subtyp nach Lauren, Anwendung einer adjuvanten Therapie und Alter umfasste, leicht höher aus (HR 4,55; 95%-KI 1,09–19,04; p = 0,04). Die FISH-Analysen zeigten überdies, dass verschiedene Tumorzellklone, die sich in den Kopienzahlvariationen zahlreicher relevanter Onkogene unterscheiden konnten, im Gewebe teilweise weniger als einen Millimeter auseinander lagen.Quelle: Chao J et al. JAMA Netw Open 2020; 3: e203652; DOI: 10.1001/jamanetworkopen.2020.3652