Neurofibromatose Klassendenken bei Typ 1
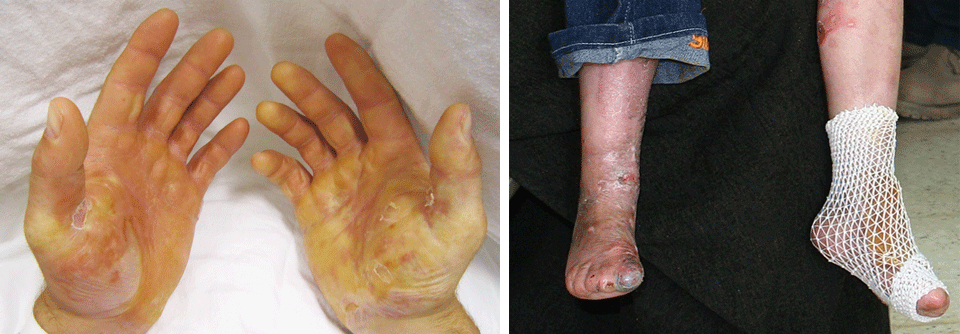
 Nuerofibrome sind gutartige Hautknoten, die von den Umhüllungen der peripheren Nerven ausgehen.
© chiew – stock.adobe.com
Nuerofibrome sind gutartige Hautknoten, die von den Umhüllungen der peripheren Nerven ausgehen.
© chiew – stock.adobe.com
Auslöser der Neurofibromatose Typ 1 (NF1) sind autosomal dominant vererbte Mutationen im Tumorsuppressorgen NF1. Bekannt sind mehr als 1.400 pathogene Varianten. Prinzipiell kann sich die NF1 als Multisystemerkrankung kutan, neurologisch, ophthalmologisch und orthopädisch manifestieren, erläutern Dr. Christina Bergqvist vom Hôpital Henri-Mondor in Créteil und Kollegen. Als typisch gelten Neurofibrome, also gutartige, von den Umhüllungen peripherer Nerven ausgehende Hautknoten, multiple Café-au-lait-Flecken, eine sommersprossenartige axilläre oder inguinale Pigmentierung („Freckling“), benigne Irishamartome (Lisch-Knötchen) sowie Lernschwäche und Skoliose. Der Phänotyp ist sehr variabel und durch zusätzliche Einflussfaktoren so komplex, dass die Prognoseeinschätzung selbst zwischen Familienmitgliedern mit identischer NF1-Mutation schwierig ist.
Die Dermatologin und ihre Kollegen haben klinische Phänotypen identifiziert, welche sich möglicherweise bestimmten Kofaktoren zuordnen lassen. Das empirisch entwickelte Klassifikationsschema basiert auf den Daten von 1.351 NF1-Patienten im Alter zwischen 17 und 81 Jahren. Über die typischen klinischen Beobachtungen ließen sie sich in drei verschiedene Subklassen aufteilen.
52 % der Betroffenen bildeten die Klasse I („kutane Neurofibrome“), deren namensgebende Hauptkennzeichen subkutane Nerventumoren darstellen. Außerdem waren plexiforme Neurofibrome und blau-rote Hautflecken (als Variante der kutanen Neoplasien) häufig. Diese klassische, meist mild verlaufende Form schien außerdem mit der Entwicklung einer Skoliose assoziiert (relatives Risiko 1,7).
Weitere 44 % der Patienten – betroffen waren überproportional häufig Männer (RR 1,4) – bildeten die Klasse II („subkutane Neurofibrome“), die sich durch mindestens zehn subkutane Nerventumoren auszeichnete und gehäuft Personen mit familiärer NF1-Belastung umfasste. Anders als in bisherigen Untersuchungen zeigte sich bei der vermehrt subkutanen Manifestation im Vergleich zur Klasse I allerdings kein erhöhtes Risiko für interne Neurofibrome.
Die übrigen rund 4 % der Patienten bildeten die Klasse III, welche NF1-Kranke mit dysmorphem Phänotyp sowie Lernstörungen umfasste. Diese schwere Verlaufsform ging mit einem rund fünffach erhöhten Risiko für Sehbahn-Gliome bzw. eine Epilepsie einher. Bereits beschriebene Eigenschaften, die dieser Gruppe zugeordnet werden, umfassen neben dem niedrigeren IQ beispielsweise auch ungewöhnliche Gesichtsmerkmale inkl. -deformitäten und ein erhöhtes Risiko für maligne periphere Nervenscheidentumoren. Außerdem entwickeln diese Patienten schon jung Neurofibrome.
Eine große Stärke ihres vorgestellten Systems sei, dass es primär auf klinischen und für Dermatologen einfach zu erfassenden Kriterien beruhe, resümieren Dr. Bergqvist und Kollegen. Dennoch hänge eine solche Einteilung immer von der Vorauswahl der eingeschlossenen Kriterien ab. Ohne weitere größere Kohortenstudien lassen sich daher keine Aussagen zur Generalisierbarkeit treffen. Außerdem gilt es zu klären, ob den beschriebenen Phänotypklassen spezifische genetische Konstellationen zugrunde liegen.
Quelle: Bergqvist C et al. J Eur Acad Dermatol Venereol 2022; DOI: 10.1111/jdv.17974