
Sandsturm in der Lunge
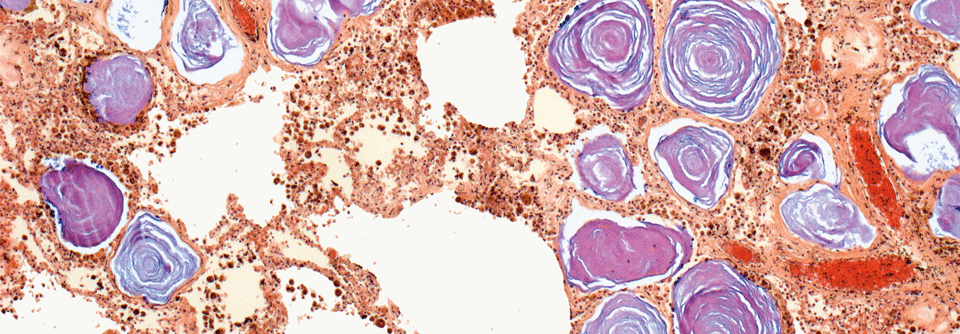 Lungengewebe eines PAM-Patienten unter dem Lichtmikroskop: In den Alveolen stellen sich Kalziumphosphatkristalle (lila) dar.
© Science Photo Library/Gschmeissner, Steve
Lungengewebe eines PAM-Patienten unter dem Lichtmikroskop: In den Alveolen stellen sich Kalziumphosphatkristalle (lila) dar.
© Science Photo Library/Gschmeissner, Steve
Die PAM entsteht durch Mutation des Natrium-Phosphat-Transportproteins NPT2B, in deren Folge es zur Ansammlung winziger Kalziumphosphatkristalle im Alveolarlumen kommt, erklärte Prof.Dr. Dirk Koschel, Chef der Pneumologie am Universitätsklinikum Dresden und am Fachkrankenhaus Coswig. Weltweit sind bisher ca. 1.000 Fälle beschrieben. Die PAM kann in jedem Alter auftreten, wird aber meist in der zweiten oder dritten Lebensdekade manifest. Die klinischen Symptome sind unspezifisch. „Oft besteht eine Diskrepanz zwischen der geringen Symptomatik und dem ausgeprägten radiologischen Befund“, so Prof. Koschel. Extrapulmonale Manifestationen kommen sehr selten vor.
Radiologisch unterscheidet man vier Stadien:
- geringer Befall mit wenigen Mikrolithen
- das typische, an einen Sandsturm erinnernde Bild mit Betonung der Mittel- und Unterfelder
- die zunehmende Dissemination mit interstitieller Septenverdickung
- extensive Kalkablagerungen („weiße Lunge“) mit Fibrosierungen und Mikrozysten, manchmal mit Pleurabeteiligung
Kalkablagerungen in der Lunge können auch bei anderen Erkrankungen entstehen, etwa bei Fibrosen oder Metastasierungen in die Lunge, die als Differenzialdiagnosen ausgeschlossen werden müssen. „Aber dieses radiologische Bild ist schon ziemlich eindeutig, vor allem wenn es in jungen Jahren auftritt“, sagte der Pneumologe.
Mikrolithensuche in Biopsie, Lavage oder Sputum
Der diagnostische Algorithmus sieht nach dem Röntgen und ergänzender HRCT die Genanalyse vor. Wird eine der inzwischen über 30 bekannten Mutationen im SLC34A2-Gen nachgewiesen, gilt dies als Beweis. Bei negativer Genanalyse muss in der Lungenbiopsie, alternativ in der bronchoalveolären Lavage oder im Sputum nach Mikrolithen gesucht werden.
Therapeutisch stellt die PAM eine Herausforderung dar, verschiedene Strategien sind bereits versucht worden: Kortison, Bisphosphonate, Kalziumbinder und Ganzlungen-Lavage. Einen durchschlagenden Erfolg hat keine gebracht. Kortison und Bisphosphonate, die bei anderen kalzifizierenden Erkrankungen gute Effekte zeigen, wirken praktisch gar nicht. Zwischenzeitlich hatten Pneumologen auf die Lungentransplantation gesetzt, „aber es gibt Berichte, dass in der transplantierten Lunge wieder eine PAM aufgetreten ist“, so Prof. Koschel. Die Hoffnungen ruhen jetzt auf Gentherapien, welche die Funktion von NPT2B wiederherstellen sollen.
Noch seltener als PAM kommt SAVI vor, die STING-associated Vasculopathy with Onset in Infancy. STING steht dabei für Stimulation of INterferon Genes. Weltweit gibt es rund 70 betroffene Patienten aus 50 Familien – eine davon lebt in Dresden und fiel den dortigen Ärzten schon 2002 auf, zwölf Jahre vor der Erstbeschreibung des Krankheitsbildes im „New England Journal of Medicine“. Erkankt waren Mutter, Tochter und Sohn: Alle litten an einer seropositiven Rheumatoiden Arthritis, begleitet von einer Vaskulitis der kleinen Gefäße, die zu Lungenfibrose, Glomerulonephritis und Hautläsionen an den Akren führte. 2009 wurde der Sohn erneut wegen einer interstitiellen Lungenerkrankung (ILD) behandelt. Mutter und Schwester waren da bereits gestorben: die Mutter mit 35, die Schwester mit 19 Jahren.
Anfang 2022 nahm man den 2011 geborenen Enkel wegen einer ILD schwerst krank in die Kinderklinik auf. Neben den Lungenbefunden mit Milchglas und zwei zystischen Läsionen im Lungenröntgenbild hatte er eine Vaskulopathie an Händen und Füßen mit frostbeulenähnlichen Läsionen und eine schwere Glomerulonephritis mit sekundärer Hypertonie. Der Vater des Kindes wies ebenfalls eine ganze Reihe schwerer Begleiterkrankungen auf: massive pulmonale Hypertonie, Herzinsuffizienz, COPD und ähnliche Chilblain-Läsionen an den Akren.
Die Biopsie zeigte eine Dermatitis mit tiefer perivaskulärer und periadnexieller Entzündung bei starker Typ-I-IFN-Aktivierung. Die daraufhin veranlasste genetische Analyse ergab die für SAVI typische gain-of- Function-Mutation im STIN1-Protein. Diese verursacht eine Proteindysfunktion im Transport von Stoffen zwischen endoplasmatischem Retikulum und Golgi-Apparat. Die resultierende Fehlfunktion des angeborenen Immunsystems führt zu Autoimmunität (rheumatoide Arthritis) und Autoinflammation. „Patienten mit SAVI haben oft eine schwere ILD mit Lungenfibrose“, berichtete Prof. Koschel. Oft finden sich wie bei dieser Familie Autoimmunkrankheiten wie die rheumatoide Arthritis, Vaskulopathien und Myositiden, Nieren- oder Leberbeteiligung sind möglich.
Therapie an Non-Adhärenz gescheitert
Wie bei vielen Orphan Diseases gestaltet sich die Therapie schwierig. SAVI spricht kaum auf Immunsuppressiva an, aber ein Therapieversuch mit JAK-Inhibitoren, die als effektiv gegen Interferon-assoziierte Pathologien gelten, kann sich lohnen. Bei Vater und Sohn waren die Erfolgschancen allerdings mäßig, weil JAK-Inhibitoren kaum auf die Lungenbefunde wirken. Die Dresdener Kollegen begannen bei dem Vater eine Behandlung mit Nintendanib unter der Vorstellung, dass Antifibrotika beim PF-ILD-Phänotyp etwas ausrichten könnten. Ob das funktioniert hat, lässt sich nicht beurteilen, weil „sich der Patient nach zwei Monaten der Behandlung entzog“, wie Prof. Koschel berichtete.
Kongressbericht: 9. Symposium Seltene Lungenerkrankungen im virtuellen Fokus
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).
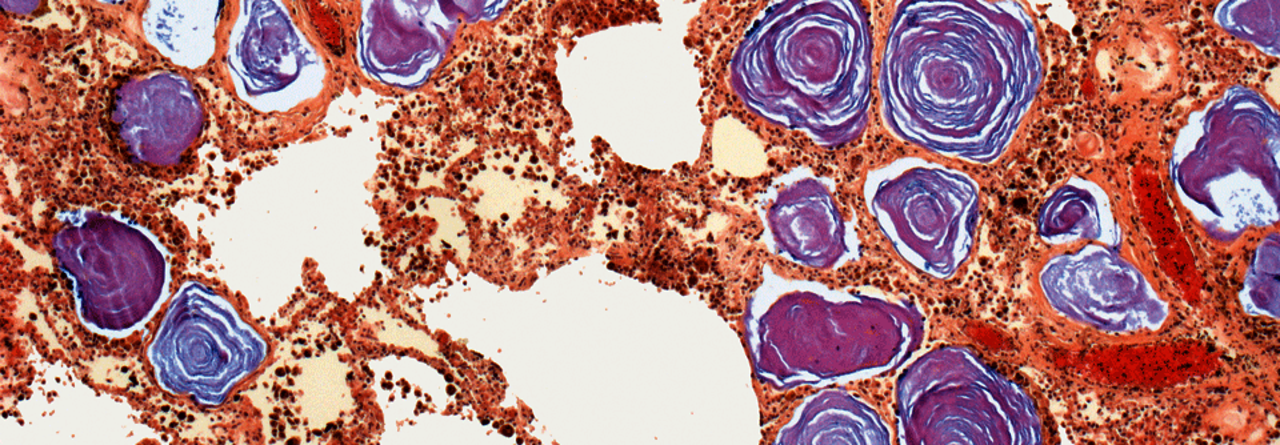 Lungengewebe eines PAM-Patienten unter dem Lichtmikroskop: In den Alveolen stellen sich Kalziumphosphatkristalle (lila) dar.
© Science Photo Library/Gschmeissner, Steve
Lungengewebe eines PAM-Patienten unter dem Lichtmikroskop: In den Alveolen stellen sich Kalziumphosphatkristalle (lila) dar.
© Science Photo Library/Gschmeissner, Steve