Pulmonale alveoläre Mikrolithiasis mit Lungentransplantation behandeln?
 Bei der PAM akkumulieren Mikrolithen aus Hydroxyapatit im Lumen von Alveolen.
© wikipedia/Dr. Mikael Häggström (CC0 1.0)
Bei der PAM akkumulieren Mikrolithen aus Hydroxyapatit im Lumen von Alveolen.
© wikipedia/Dr. Mikael Häggström (CC0 1.0)
Die hereditäre pulmonale alveoläre Mikrolithiasis (PAM) ist gekennzeichnet durch eine Akkumulation von knochenähnlichen Hydroxyapatit-Mikrolithen im Lumen von Alveolen. Sie äußert sich in einer langsam, über Dekaden zunehmenden respiratorischen Insuffizienz, die sich dann im mittleren Lebensalter manifestiert. Bisher wurden weltweit nur gut 1000 Fälle dieser Krankheit berichtet, schreiben Dr. Patrick Kosciuk von der Division of Pulmonary, Critical Care and Sleep Medicine der University of Cincinnati und Kollegen.
Autosomal-rezessiver Erbgang vermutet
Genetische Analysen haben als Ursache verschiedene meist homozygote Mutationen des Gens SLC34A2 identifiziert, das für den Natriumphosphat-Cotransporter NPT2B kodiert. Familienstudien lassen einen autosomal rezessiven Erbgang vermuten. Die Mutationen bedingen eine Fehlfunktion von NPT2B.
NPT2B wird vor allem im Alveolarepithel stark exprimiert und hat dort die Aufgabe, Phosphat zu exportieren, das beim Katabolismus von Surfactant-Phospholipiden durch Alveolarmakrophagen entstanden ist. Auch das Dünndarmepithel exprimiert viel NPT2B, wo es der Absorption von Phosphat aus der Nahrung dient. Eine Fehlfunktion macht sich dort aber meist nicht bemerkbar, weil renale Mechanismen die Phosphat-Homöostase aufrechterhalten.
Es drohen pulmonale Hypertonie und Fibrose
In den Alveolarräumen der Lunge lagern sich dagegen Hydroxyapatit-Mikrolithen ab, wenn NPT2B nicht funktioniert. Sie fallen im Röntgenbild und CT als mikronoduläre hyperdense Infiltrate auf. Die CT gilt dabei als aussagekräftigstes bildgebendes Verfahren. Nicht selten sieht man auch eine Beteiligung des Parenchyms mit subpleuralen interstitiellen oder interlobären septalen Verdickungen ähnlich der Lungenfibrose. Die radiologischen Veränderungen sind meist ausgeprägter, als klinische Symptome wie Belastungsdyspnoe oder trockener Husten vermuten lassen. Mit der Zeit entwickeln viele Patienten eine pulmonale Hypertonie oder Fibrose.
Lungenfunktionstests fallen in frühen Stadien noch normal aus. Später zeigt sich ein restriktives Defizit mit einer Abnahme der Diffusionskapazität für Kohlenmonoxid. Routine-Labortests ergeben meist normale Befunde. Dagegen steigt die Konzentration von Surfactant-Protein-D mit Progression der Krankheit immer stärker an.
Zum natürlichen Verlauf der PAM kann man nicht viel sagen, weil es in den wenigsten Fällen frühere Röntgenbilder gibt, die zum Vergleich herangezogen werden können. Doch Familienstudien mit mehreren Betroffenen weisen auf einen sehr variablen Verlauf hin. Es gibt Patienten, die schon früh eine Lungentransplantation brauchen und andere, die ein höheres Lebensalter weitgehend ohne klinische oder radiographische Progression erreichen. Liegen im Röntgenbild oder der CT typische Veränderungen vor, wird ein Blick auf die Familienanamnese geworfen. Gibt es dort nahe Verwandte mit genetisch gesicherter PAM, genügt das für eine definitive Diagnose. Ansonsten erfolgt die genetische Testung am Indexpatienten. Steht diese Option nicht zur Verfügung, kann man nach Mikrolithen mit einer typischen lamellären Struktur im Sputum oder Material aus der Bronchiallavage suchen.
Im Zweifel transbronchiale Biopsie anstreben
Wenn sich hier keine Mikrolithen finden, sollte eine transbronchiale Zangenbiopsie angeschlossen werden. Die videoassistierte thorakoskopische Biopsie bleibt Fällen vorbehalten, in denen weniger invasive Verfahren nicht erfolgreich waren oder nicht zur Verfügung stehen.
Präklinisch haben sich bereits Ansätze für eine mögliche Therapie herauskristallisiert, z.B. die genetische Korrektur der NPT2B-Expression im Alveolarepithel. Im Tiermodell ließ sich durch eine extrem phosphatarme Diät und eine therapeutische Lavage mit Calciumchelatoren die Bildung von Mikrolithen verringern.
Die meisten Therapieversuche haben enttäuscht
Sich phosphatarm zu ernähren, kann man Patienten durchaus raten, wenngleich sich die Phosphatrestriktion in einem beim Menschen machbaren Ausmaß bisher nicht positiv auf die Progression der Erkrankung ausgewirkt hat. Der Calciumchelator Natriumthiosulfat erwies sich in Einzelfällen als erfolgreich. Die meisten empirischen Therapieversuche haben jedoch enttäuscht, z.B. Bisphosphonate, inhalative oder systemische Steroide.
Auch eine Ganzlungenlavage hatte bei Patienten nicht den erhofften Effekt auf die radiographischen Befunde oder klinischen Symptome, obwohl damit eine Menge kleinerer solider Partikel entfernt wurden – die in den Alveolarräumen abgelagerten aber offenbar nicht. Die einzige wirklich erfolgreiche Therapie ist die Lungentransplantation. Bisher gab es keinen Hinweis auf rezidivierende Mikrolithenbildung bei den so behandelten PAM-Patienten.
Quelle: Kosciuk P et al. Eur Resp Rev 2020; 29: 200024; DOI: 10.1183/16000617.0024-2020
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).

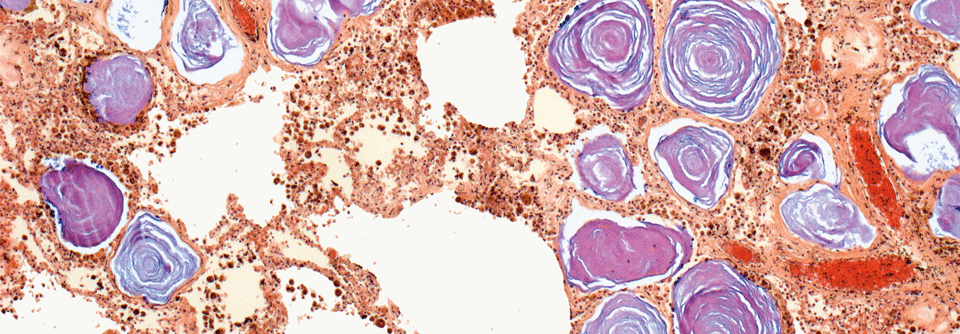
 Bei der PAM akkumulieren Mikrolithen aus Hydroxyapatit im Lumen von Alveolen.
© wikipedia/Dr. Mikael Häggström (CC0 1.0)
Bei der PAM akkumulieren Mikrolithen aus Hydroxyapatit im Lumen von Alveolen.
© wikipedia/Dr. Mikael Häggström (CC0 1.0)