Stillstand in den kleinen Gefäßen
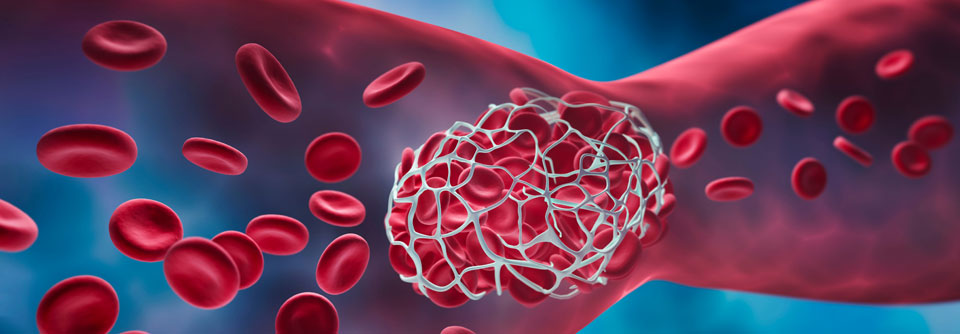 Eine gefährliche Form der thrombotischen Mikroangiopathie ist die thrombotisch-thrombozytopenische Purpura, kurz TTP.
© peterschreiber.media – stock.adobe.com
Eine gefährliche Form der thrombotischen Mikroangiopathie ist die thrombotisch-thrombozytopenische Purpura, kurz TTP.
© peterschreiber.media – stock.adobe.com
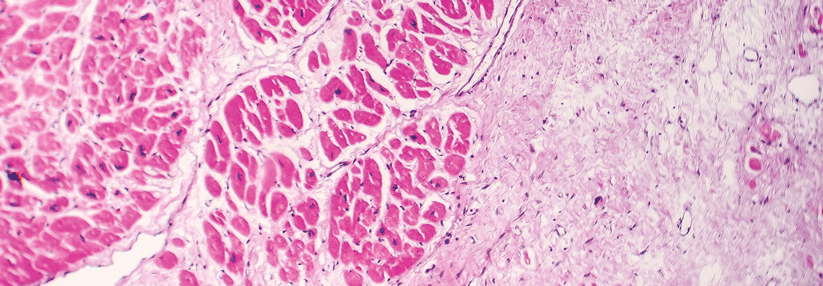
Mit dem Begriff der thrombotischen Mikroangiopathie (TMA) beschreibt man eine Gruppe von Erkrankungen, bei denen es in den kleinen Gefäßen zu Endothelschäden und Gefäßokklusionen kommt. Häufig ist das mit einer intraluminalen Thrombusbildung verbunden. Die mikroangiopathische hämolytische Anämie beruht auf einer mechanischen Fragmentierung von Erythrozyten, die durch verschlossene Gefäße zirkulieren. Es kommt zu Ischämie und Inflammation, die das Gewebe angreifen und schließlich zu Schäden an den Organen und der Gefahr eines Multiorganversagens führen, schreibt das Team um Joshua Leisring vom Ohio State University Wexner Medical Center in Columbus.
Die thrombotisch-thrombozytopenische Purpura und das hämolytisch-urämische Syndrom zählen zu den primären TMA-Syndromen. Im klinischen Alltag beobachtet man die thrombotische Mikroangiopathie häufiger bei Schwangeren oder im Zusammenhang mit Grundleiden wie einer autoimmunen Erkrankung und Infektionen. Unabhängig von der Ursache geht die TMA mit erheblicher Morbidität und Mortalität einher, weshalb rasches Handeln geboten ist, betonen die Autoren.
Der thrombotisch-thrombozytopenischen Purpura (TTP) liegt eine reduzierte Aktivität des Plasmaenzyms ADAMTS13 zugrunde. Dieses Enzym spaltet die Multimere des von-Willebrand-Faktors (vWF), die von Gefäßendothelzellen sezerniert werden. Bleibt die Spaltung aus, triggern ultralange Multimere eine übermäßige Thrombozytenaggregation. In der Konsequenz bilden sich Thromben in Arteriolen und Kapillaren. In mehr als 90 % der Fälle handelt es sich um eine erworbene immunvermittelte TTP mit Autoantikörpern gegen ADAMTS13. Eine kongenitale thrombotisch-thrombozytopenische Purpura findet sich häufiger bei Kindern mit ADAMTS13-Mutationen.
Ohne Therapie überlebt kaum einer der Patienten
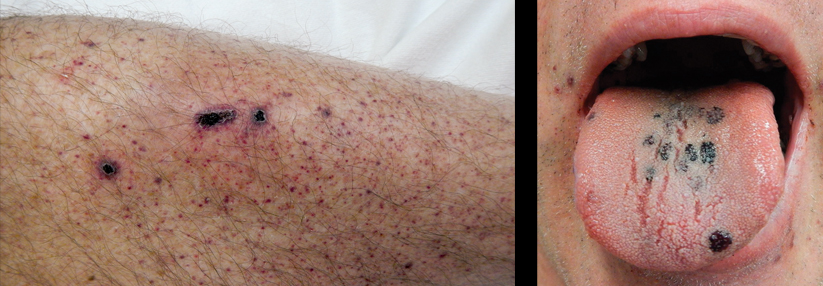
Die TTP gilt als hämatologischer Notfall, unbehandelt führt sie bei mehr als neun von zehn Betroffenen zum Tod. Allerdings sieht man nur bei wenigen Patienten die klassische Kombination aus Thrombozytopenie, mikroangiopathischer hämolytischer Anämie, neurologischen Veränderungen, Nierenfunktionsstörung und Fieber.
Im Vergleich zu anderen Formen der thrombotischen Mikroangiopathie führt die TTP häufig zu einer ausgeprägteren Thrombozytopenie und milderer Nierenfunktionsstörung. Thrombozytenwerte < 30 × 109/l und ein Kreatinin < 2,26 mg/dl bei klinischem Verdacht sprechen für eine TTP. Eine ADAMTS13-Aktivität < 10 IU/dl untermauert die Diagnose, bei einer Aktivität > 20 IU/dl sollte an andere Ursachen gedacht werden.
Grundpfeiler der TTP-Therapie ist der Plasmaaustausch, um intaktes ADMTS13 zuzuführen und Autoantikörper zu entfernen. Bei angeborenem Enzymmangel können Plasmainfusionen zum Ausgleich des Defizits ausreichen. Liegt eine immunvermittelte TTP vor, empfehlen die Autoren zusätzlich Kortikosteroide und Rituximab, um die Produktion von Autoantikörpern zu drosseln. Das humanisierte Anti-vWF-Immunglobulinfragment Caplacizumab hemmt die Interaktion zwischen den von-Willebrand-Faktor-Multimeren und Thrombozyten. Mit ihm gelingt es, frühe Krankheitsrezidive zu verhindern.
HUS wird oft durch Shigellen oder E. coli ausgelöst
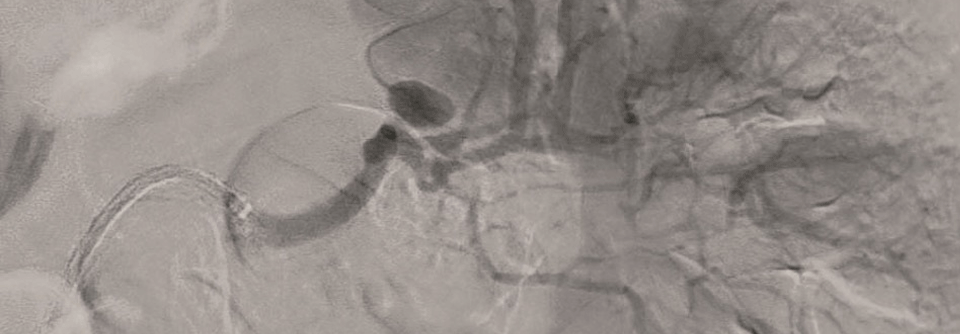
Das typischerweise durch shigatoxinbildende Erreger verursachte hämolytisch-urämische Syndrom (HUS) wurde initial „nur“ als thrombotische Mikroangiopathie mit der klassischen Trias Thrombozytopenie, mikroangiopathische hämolytische Anämie sowie schwere Nierenfunktionsstörung charakterisiert. Noch immer ist es die häufigste TMA im Kindesalter, tritt aber auch bei Erwachsenen auf. Häufig liegt der Erkrankung eine Infektion mit enterohämorrhagischen Escherichia coli, aber auch mit Shigella dysenteriae zugrunde. Die Therapie erfolgt supportiv mit Hydratation und Erythrozytentransfusionen nach Bedarf.
Das komplementvermittelte HUS (CM-HUS) gilt als atypische Form des Syndroms. Ursache sind meist genetisch bedingte Veränderungen im Komplementsystem. Für die Behandlung des CM-HUS stehen Eculizumab und Ravulizumab zur Verfügung, die beide den Komplementfaktor C5 blockieren.
Eine Therapie mit C5-Inhibitoren sollte man schon beim klinischen Verdacht auf ein CM-HUS und vor Eintreffen der entsprechenden Laborbefunde erwägen. Wird die Therapie mit Eculizumab innerhalb einer Woche nach Symptombeginn eingeleitet, sind die Langzeitergebnisse mit Blick auf die Nierenfunktion oft besser. Man muss bedenken, dass die C5-Inhibitoren teuer sind und das Infektionsrisiko erhöhen. Bei TMA in der Schwangerschaft oder im Zuge anderer Erkrankungen richtet sich die Therapie nach dem Grundleiden und der klinischen Präsentation der Mikroangiopathie.
Quelle: Leisring J et al. Arthritis & Rheumatology 2023; DOI: 10.1002/art.42681
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).
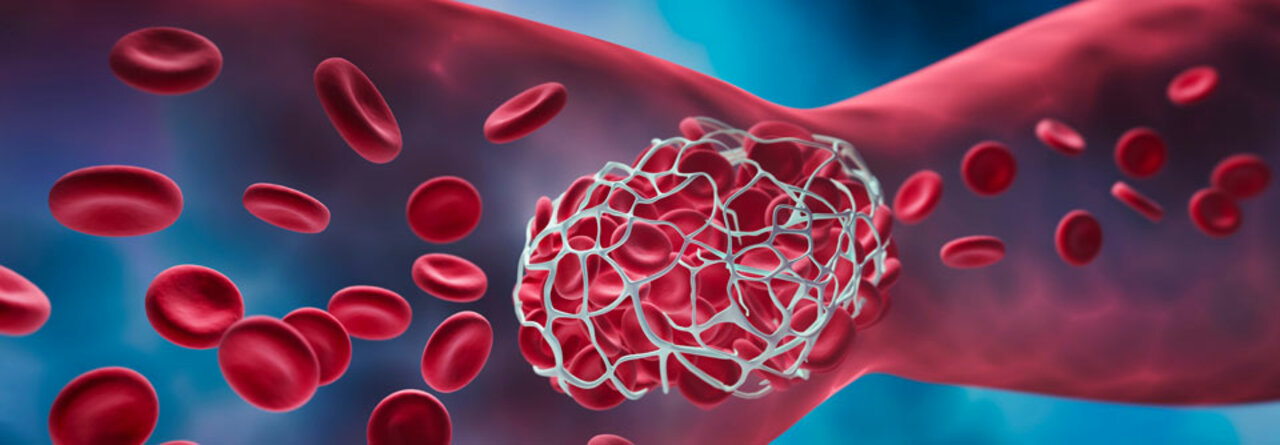 Eine gefährliche Form der thrombotischen Mikroangiopathie ist die thrombotisch-thrombozytopenische Purpura, kurz TTP.
© peterschreiber.media – stock.adobe.com
Eine gefährliche Form der thrombotischen Mikroangiopathie ist die thrombotisch-thrombozytopenische Purpura, kurz TTP.
© peterschreiber.media – stock.adobe.com