
Zur Not off label ans Ziel kommen
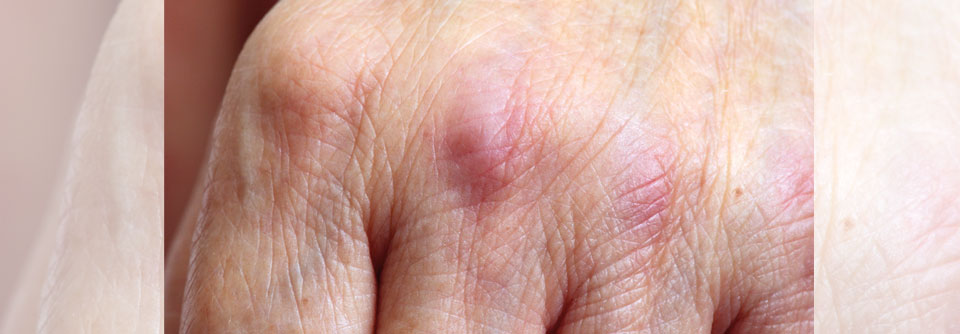 Flache, rötlich-livide Papeln und Plaques unterschiedlicher Größe auf den Streckseiten von Händen und Fingern. Diese Gottron-Papeln sind typisch für die Dermatomyositis und kommen sowohl bei Erwachsenen als auch bei Kindern vor.
© Science Photo Library
Flache, rötlich-livide Papeln und Plaques unterschiedlicher Größe auf den Streckseiten von Händen und Fingern. Diese Gottron-Papeln sind typisch für die Dermatomyositis und kommen sowohl bei Erwachsenen als auch bei Kindern vor.
© Science Photo Library
Die juvenile Dermatomyositis (JDM) ist eine äußerst seltene Erkrankung. So gab es laut der Kinderquerschnittuntersuchung 2014–2016 nur 196 betroffene Patienten in Deutschland. Dennoch ist die JDM hochrelevant, betonte Dr. Claas Hinze, Kinderrheumatologe an der Universitätsklinik Münster. Denn ihre meist langjährigen Verläufe können zu ausgeprägten Spätfolgen führen. Dazu gehören nicht nur Kalzinosen und Kontrakturen. Besonders gefürchtet sind auch partielle oder generelle Lipodystrophien, die schwerwiegende metabolische Folgen nach sich ziehen.
Adulte und juvenile Form vermutlich zwei Entitäten
Die juvenile und die adulte Dermatomyositis sind wahrscheinlich keine identischen Erkrankungen. Das Alter bei Beginn der JDM liegt zwischen fünf und zehn Jahren, das bei der adulten Dermatomyositis zwischen 50 und 60. Im Unterschied zur adulten Form überwiegt bei den betroffenen Kindern die Dermatomyositis, andere Ausprägungen wie die Polymyositis sind bei ihnen nur selten zu sehen. Paraneoplasien kommen bei der JDM praktisch nie vor, bei den Erwachsenen in etwa 10 % der Fälle. Kinder entwickeln dagegen häufiger Kalzinosen (40 % vs. 20 %), zudem drohen bei ihnen Wachstumsverzögerung und verspätete Pubertät.
Hintergrund der Erkrankung ist ein autoimmuner Teufelskreis, der mit der Aktivierung und Infiltration von Entzündungszellen im Gewebe beginnt. Die dadurch ausgelösten Interferonsignale führen zu Schäden im Muskelgewebe, in der Haut und an den Gefäßen. Bei der Regeneration der Muskelfasern kommt es vermutlich zu einer Überexpression muskelspezifischer Antigene, erklärte Dr. Hinze. Diese werden dann durch myositisspezifische Autoantikörper erkannt, worauf es zur lokalen Komplementaktivierung und zur erneuten Aktivierung und Infiltration von Entzündungszellen kommt – und das Ganze von vorne losgeht.
JDM und Interferon
Typ-1-Interferon spielt beim autoimmun-entzündlichen Geschehen der JDM eine wichtige Rolle. So ist der Interferonspiegel im peripheren Blut ähnlich hoch wie bei monogenetischen Interferonopathien oder beim systemischen Lupus erythematodes. Zudem korreliert die Typ-1-Interferon-Signatur im Blut mit der Krankheitsaktivität – hohe Werte bedeuten eine hohe Aktivität. Unter einer effektiven Therapie normalisieren sich die Typ-1-Interferonspiegel wieder.
Die Diagnose erfolgt nach den modifizierten Kriterien nach Bohan und Peter, unterstützt durch supportive Befunde. Hauptkriterium sind die typischen Hautveränderungen. Je nachdem, wie viele der folgenden vier Punkte dazukommen, spricht man von einer möglichen (ein Kriterium), wahrscheinlichen (zwei Kriterien) oder definitiven (ab drei Kriterien) JDM:
- proximale Muskelschwäche
- erhöhte Muskelenzyme
- typische MRT-Befunde
- typische histopathologische Befunde
Myositisspezifische Antikörper bei der JDM
Die myositisspezifischen Antikörper der JDM unterscheiden sich von denen der adulten Form. Während dort die Anti-Synthetase vorwiegt, sieht man bei den Kindern am häufigsten die Anti-NXP2-positive JDM. Diese ist durch ausgeprägte Kalzinosen und Chronizität charakterisiert. Am zweithäufigsten finden sich Anti-TIF1-gamma-Antikörper, bei dieser Form der JDM sind Vaskulopathien und Photosensitivität besonders ausgeprägt. Am dritthäufigsten kommen bei der JDM Anti-MDA5-Antikörper vor. Sie werden vermehrt von Lungenbeteiligungen und anderen extramuskulären Manifestationen begleitet.
Als Behandlungsstrategie wird neben den Basismaßnahmen (effektiver Sonnenschutz, Physiotherapie, Sport, ggf. Vitamin D, Kalzium und Hydrochloroquin) ein Treat-to-Target-Ansatz mit dem Ziel der Remission empfohlen. Zur Messung von Krankheitsaktivität und Therapieerfolg gibt es neben der Bestimmung der Muskelenzyme verschiedene validierte Tools, z.B. die Childhood Myositis Assessment Scale, das Myositis Disease Activity Assessment Tool oder der Juvenile Dermatomyositis Activity Index. Die Lebensqualität lässt sich mit dem Childhood Health Assessment Questionnaire erfassen, das Ausmaß einer Verbesserung mithilfe des ACR/EULAR-Verbesserungs-Scores. Zudem existieren Definitionen für eine inaktive Erkrankung.
JAK-Inhibitoren besonders vielversprechend
Die Behandlung erfolgt gemäß klinischer Erfahrung und nationalen und internationalen Konsensus-Therapieplänen. Als Induktionstherapie werden Glukokortikoide und eine antirheumatische Basistherapie mit Methotrexat, Azathioprin, Ciclosporin A oder Mycophenolat-Mofetil empfohlen. Zugelassen für die Dermatomyositis sind allerdings nur Glukokortikoide, Azathioprin und Immunglobuline. Optional sind bei schwerer JDM auch Immunglobuline i.v. möglich. Ziel ist eine inaktive Erkrankung nach zwölf Monaten. Dann können die (vorher evtl. schon reduzierten) Glukokortikoide ganz abgesetzt werden, die anderen Therapien werden fortgeführt. Wird das Ziel nicht erreicht, kommen zusätzlich weitere glukokortikoidsparende Wirkstoffe infrage. Davon ist jedoch keiner für diese Indikation zugelassen. Neben den Basistherapeutika handelt es sich dabei um Rituximab, Tacrolimus, TNF-α-Blocker oder JAK-Inhibitoren. Letztere sind dabei in Bezug auf die Wirksamkeit besonders ermutigend, berichtete Dr. Hinze. In Fallberichten und Open-Label-Studien mit insgesamt 23 Patienten führten sie in 70 % der Fälle zu einer mindestens moderaten Verbesserung der Symptomatik.Quelle: Deutscher Rheumatologiekongress 2021 – virtuell
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).

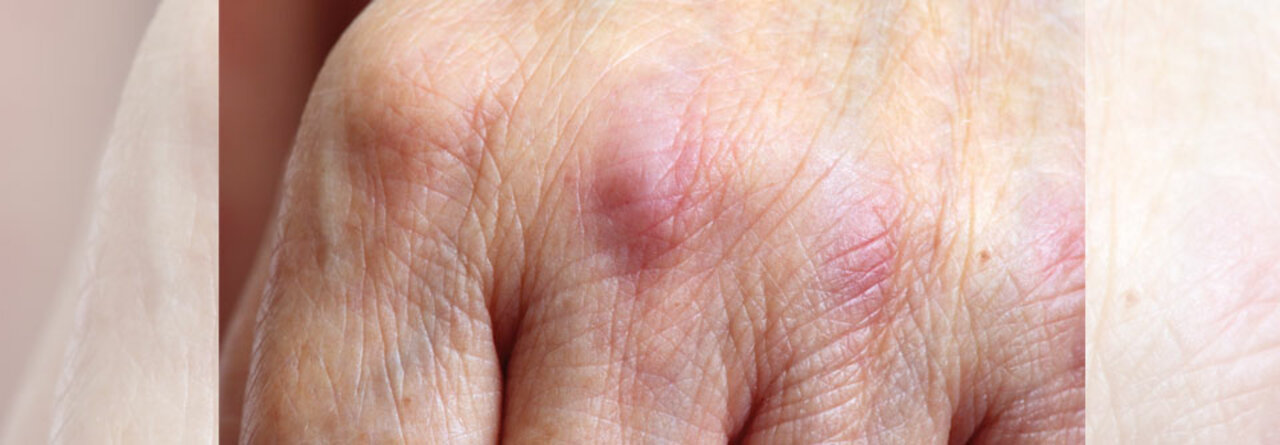 Flache, rötlich-livide Papeln und Plaques unterschiedlicher Größe auf den Streckseiten von Händen und Fingern. Diese Gottron-Papeln sind typisch für die Dermatomyositis und kommen sowohl bei Erwachsenen als auch bei Kindern vor.
© Science Photo Library
Flache, rötlich-livide Papeln und Plaques unterschiedlicher Größe auf den Streckseiten von Händen und Fingern. Diese Gottron-Papeln sind typisch für die Dermatomyositis und kommen sowohl bei Erwachsenen als auch bei Kindern vor.
© Science Photo Library