Interstitielle Lungenerkrankungen Früherkennung muss aufs Tapet
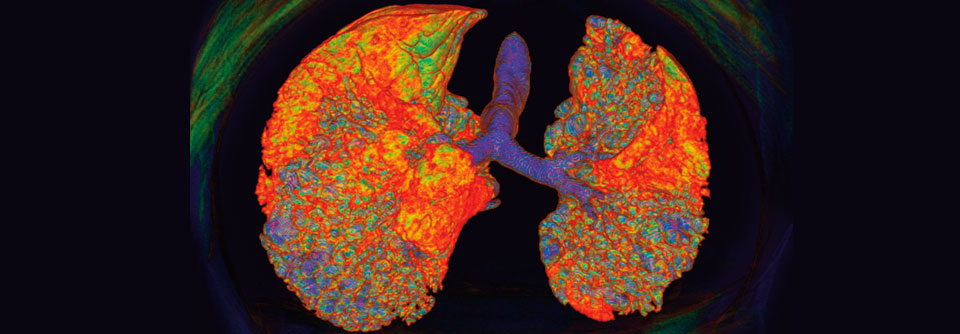
 Entwickelt ein Patient eine interstitielle Lungenerkrankung wie hier im 3D-CT erkennbar, sollte man seine Verwandten ersten Grades auf Lungenveränderungen screenen.
© Science Photo Library/Fung, K.H.
Entwickelt ein Patient eine interstitielle Lungenerkrankung wie hier im 3D-CT erkennbar, sollte man seine Verwandten ersten Grades auf Lungenveränderungen screenen.
© Science Photo Library/Fung, K.H.
Die Prognose interstitieller Lungenerkrankungen (ILD) bleibt trotz aller therapeutischen Innovationen schlecht. Jeder zweite Patient mit idiopathischer Lungenfibrose (IPF) überlebt das fünfte Jahr nach Diagnose nicht. „Catching the Bull Before It Destroys the Shop“ hatte die American Thoracic Society deshalb das Symposium zur ILD-Frühdiagnostik betitelt. Um den ILD-Stier zu erwischen, ist es unabdingbar, alle Verwandten ersten Grades von Patienten mit familiärer, aber auch sporadischer ILD auf Anzeichen einer beginnenden pulmonalen Fibrose zu screenen, meinte Prof. Dr. Gary Hunninghake vom Brigham and Women’s Hospital, Boston. Denn bisherige klinische Studien zur antifibrotischen Therapie bei IPF zeigen, dass Patienten in frühen Stadien nicht nur ansprechen, sondern besonders stark profitieren: Wo viel FVC zu verlieren ist, lässt sich eben auch viel retten.
Alter, LuFu, Genetik und Rauchen als Risikofaktoren
Der Anteil unerkannter interstitieller Lungenabnormalitäten (ILA, s. Kasten) bei Verwandten von ILD-Patienten ist sehr hoch, betonte Prof. Hunninghake. In einer Studie, an der 105 erstgradige Verwandte von Patienten mit familiärer oder sporadischer Lungenfibrose teilnahmen, entdeckte sein Team bei jedem Dritten Zeichen einer ILA. Als Risikofaktoren fanden die Forscher Alter, reduzierte Lungenfunktion (FVC, TLC, DLCO), verminderte Telomerlänge in Lymphozyten sowie multiple Kopien der MUC5B-Promotervariante. Registerstudien kamen zu ähnlichen, etwas niedrigeren Raten und identifizierten Rauchen sowie männliches Geschlecht als weitere Risikofaktoren.
Die ILA-Definition
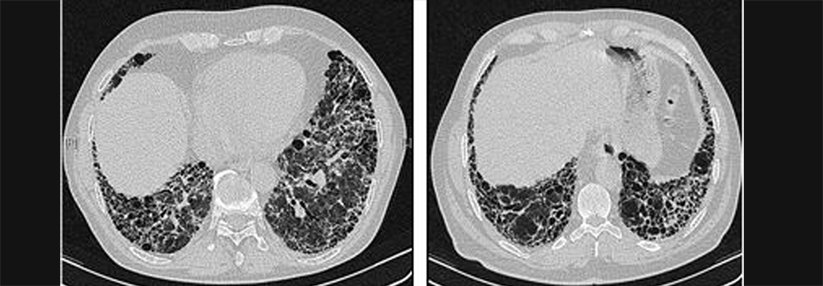
Zur Abgrenzung gegen die manifeste ILD hat die Fleischner-Society 2020 eine Definition für interstitielle Lungenabnormalitäten (ILA) vorgelegt. Demnach handelt es sich um ILA, wenn bei einem Menschen ohne ILD-Verdacht als Zufallsbefund Milchglas- oder retikuläre Veränderungen, Traktionsbronchiektasen, Honigwabenphänomene oder nicht-emphysematöse Zysten gefunden werden, die mindestens 5 % eines Lungenabschnitts betreffen.
Quelle: Hatabu H et al. Lancet Respir Med 2020; 8: 726-737; DOI: 10.1016/S2213-2600(20)30168-5
„Ich denke, es bringt nichts, die ILA-Prävalenz in Familien zu diskutieren, ohne das Alter zu berücksichtigen“, so Prof. Hunninghake. Die Inzidenz interstitieller Veränderungen steigt mit dem Alter und erreicht relevante Größenordnungen ab Mitte 50. Für Prof. Hunninghake ist dies der früheste Zeitpunkt, ab dem ein Screening per Lungenfunktionstest starten könnte, das nur bei auffälligen Befunden durch eine CT ergänzt werden sollte. Ob es eine Altersgrenze gibt, ab der die ILA-Suche enden sollte, lässt sich derzeit nicht beantworten.
Dass ein ILA-Screening Sinn macht, steht für ihn außer Frage: „Es identifiziert nicht nur auffällige Befunde – ein wesentlicher Anteil von ILA-Patienten zeigt eine progrediente Fibrose.“ Als Beleg führte er eine Auswertung der Framingham Heart Study an, die klar zeigt, dass ILA häufig fortschreiten und dass dies mit einem beschleunigten Verlust an Lungenfunktion und einem deutlich erhöhten Sterberisiko einhergeht.
Einer aktuellen, noch nicht publizierten Analyse zufolge verdoppelt sich bei vorliegenden ILA die Mortalität – wohlgemerkt bei einem zuvor unerkannten Bildgebungsbefund ohne klinische Symptome. Der Effekt auf die Sterblichkeit liegt in der gleichen Größenordnung wie der einer koronaren Herzerkrankung und vielen Krebsformen. ILA sind definitiv kein gutartiger Befund, konstatierte daher Prof. Hunninghake. In seiner eigenen Studie erwiesen sich die ILA-Befunde bei zwei von drei Angehörigen von ILD-Patienten als progredient. Fast die Hälfte erfüllten nach zwei Jahren die Definition einer progressiven Lungenfibrose, ermittelt anhand von CT-Befund und FVC-Verlust. „Wir sollten bedenken, dass es bisher kein Anzeichen gibt, dass sich eine einmal eingetretene Fibrose zurückdrehen lässt.“
Schäden im subklinischen Stadium aufspüren
Dr. Anna Podolanczuk, Weill Cornell Medicine New York, beschäftigt sich mit Bildgebungs- und Blutbiomarkern, die helfen sollen, ILD im subklinischen Stadium zu identifizieren. „Derzeit wird die Diagnose irgendwann zwischen Symptombeginn und Tod des Patienten gestellt, aber nicht wenn Remodeling und Fibrose beginnen – das ist unbefriedigend“, so die Pneumologin. Selbst in Lungen, die im Normal-CT gesund erscheinen, lassen sich mit der richtigen Technik frühe Schäden nachweisen. Eine solche Technik ist z.B. die fortgeschrittene Dichte- und Texturmessung zum Nachweis von Inflammation und Remodeling, vor allem wenn sie sich selbstlernender KI bedient. Der Anteil von Arealen mit vermehrter Dichte (HAA) als einfachster Bildgebungsbiomarker korreliert im Follow-up über zehn Jahre mit erhöhten Spiegeln von Blutbiomarkern, etwa dem der Metallomatrixproteinase(MMP)-7, aber auch mit Defiziten der Lungenfunktion. Außerdem konnte Dr. Podolanczuk zeigen, dass ein höherer HAA-Anteil Hospitalisierungen, Gesamt- und krankheitsbedingte Mortalität bei ILD-Patienten vorhersagt.
Neben MMP-7 können Monozyten einen guten, aber natürlich relativ unspezifischen Blutbiomarker für die beginnende pulmonale Fibrosierung abgeben. Pro Anstieg der Zellzahl um eine Standardabweichung steigt die Wahrscheinlichkeit, dass ILA oder eine subklinische ILD vorliegen, um etwa 20 %.
Weitere Biomarker „in progress“ sind Telomerlänge, VCAM- und ICAM-1, P-Selectin, Apolipoprotein A1 und GDF-15. Zu allen wurden Korrelationen mit ILA, Lungenfunktionsabnahme und/oder Arealen mit vermehrter Dichte gezeigt, teils sogar mit ILD-Hospitalisierungen und Tod.
„Es gibt bisher nicht den einzelnen Biomarker, mit dem sich ILA detektieren ließe“, räumte Dr. Podolanczuk ein. Es wird wohl auf eine Kombination von Markern hinauslaufen. Wenn dann noch die KI entsprechend trainiert ist, werden sich gefährdete Patienten leichter herausfischen lassen als bisher, meinte die Kollegin.
Quelle: ATS* 2022
* American Thoracic Society