Hämoglobinopathien Sichelförmig statt bikonkav unterwegs
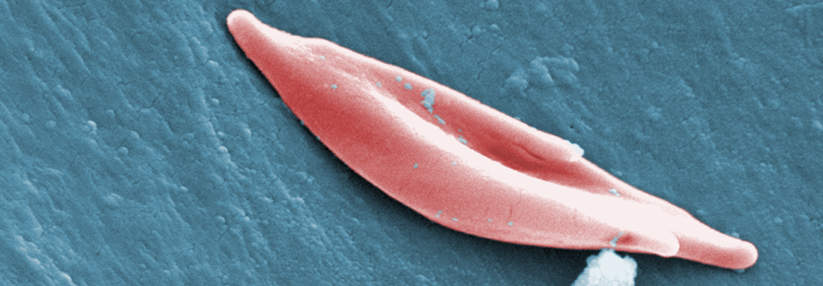
 Sichelzellen lösen sowohl akute Gefäßverschlüsse als auch chronische Organschäden aus.
© Science Photo Library/Murti, Dr. Gopal
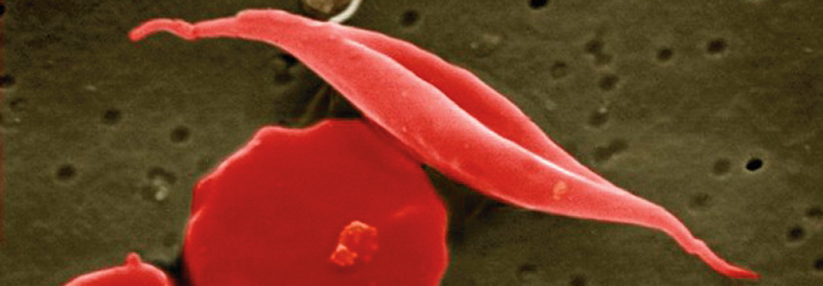
Sichelzellen lösen sowohl akute Gefäßverschlüsse als auch chronische Organschäden aus.
© Science Photo Library/Murti, Dr. Gopal
Dem pathologischen Sichelzellhämoglobin (HbS) liegt eine Punktmutation im ß-Globin-Lokus auf Chromosom 11 zugrunde. Im Gegensatz zum normalen HbA bleibt das HbS bei Deoxygenierung nicht in Lösung, sondern polymerisiert zu langen Strängen. Sie zwingen den Erythrozyten in eine Sichelform, schreibt Dr. Roswitha Dickerhoff aus München. Die verformten roten Blutkörperchen sind starr, verschlechtern die Fließeigenschaften des Blutes und haften leicht am Endothel. Dadurch kommt es zu einer chronischen Entzündung des Endothels und zu gefährlichen Gefäßreaktionen, die sich unterschiedlich manifestieren, nämlich als
- Gefäßverschlüsse mit akuten Folgen (Schmerzkrisen, Hörsturz, ZNS-Infarkte)
- chronische Organschäden (z.B. Retinopathie, Niereninsuffizienz, funktionelle Asplenie)
- Sequestrationen (Milz- oder Lebersequestration, Priapismus etc.)
Anstatt einer Vasoobstruktion oder Sequestrationen ist auch eine chronische Hämolyse möglich. Sie begünstigt u.a. Anämien oder Gallensteine.
| Manifestationen der Sichelzellkrankheiten im Erwachsenenalter | |
|---|---|
| Akute Probleme | Chronische Probleme |
| Schmerzkrisen | chronische Schmerzen |
| Sepsis (durch Pneumokokken, gramnegative Keime etc.) | avaskuläre Nekrosen (Femur, Humerus, Deckplatteneinbrüche) |
| akutes Thoraxsyndrom | Niereninsuffizienz |
| Komplikationen in der Schwangerschaft | Priapismus |
| Hörsturz (bei hoher Viskosität, HbSC) | proliferative Retinopathie (70 % bei HbSC) |
| Gallenkoliken | Alloimmunisierung, verzögerte hämolytische Transfusionsreaktion |
| Hyperbilirubinämie-Syndrom | Sichel-Hepatopathie, Zirrhose |
| Milzsequestration (HbSC, HbSLepore, HbSβ°Thal) | pulmonale Hypertonie |
| Lebersequestration | Ulzera an den Unterschenkeln |
| ZNS-Blutung | Herzinsuffizienz, Kardiomyopathie |
Die häufigste Form der Sichelzellkrankheit ist die homozygote Variante HbSS. Außerdem gibt es „compound-heterozygote“ Varianten wie HbSC, HbSD, HbSβ°Thal, HbS0Arab oder HbSLepore. Betroffene erhalten von einem Elternteil das HbS, vom zweiten eine andere β-Globin-Mutation. Sichelzellkrankheiten werden vor allem bei Immigranten aus Afrika, dem östlichen Mittelmeerraum und dem mittleren Osten beobachtet. Während früher viele Betroffene bereits im Kindesalter starben, werden heute rund 95 % erwachsen.
Funktionelle Asplenie bereits im ersten Lebensjahr
Am schwersten verlaufen HbSS, HbSβ°Thal, HbSD und HbS0Arab. Kindern mit diesen Varianten droht bereits im ersten Lebensjahr eine funktionelle Asplenie mit hoher Sepsisgefahr. Milzsequestrationen werden im Allgemeinen nur bis zum achten Lebensjahr beobachtet. Ohne entsprechende Prophylaxe bekommen etwa 12 % der Patienten bis zum 18. Lebensjahr einen ZNS-Infarkt.
Neben dem HbS ist Hämoglobin C (HbC) ein weiteres häufiges pathologisches Hämoglobin. In Kombination mit HbS entsteht die HbSC-Krankheit. Sowohl HbS als auch HbC sind in Westafrika endemisch, daher wird dort die HbSC-Krankheit recht häufig beobachtet. Bei Menschen afrikanischer Herkunft mit Mikrozytose, etwas erhöhten Retikulozyten und Schmerzsymptomatik sollte man an die Möglichkeit einer HbSC-Krankheit denken – auch wenn der Hämoglobinwert altersentsprechend ist.
Die HbSC-Krankheit unterscheidet sich deutlich von der HbSS-Krankheit:
- Kinder mit HbSC haben kein erhöhtes Risiko für eine Pneumokokken-Sepsis, sehr selten erleiden sie Schmerzkrisen oder eine Milzsequestration.
- HbSC-Betroffene erleiden im Kindesalter keine ZNS-Infarkte und nur sehr selten im Erwachsenenalter.
- Erwachsenen mit HbSC entwickeln seltener kardiale, pulmonale und renale Problemen als HbSS-Kranke.
- HbSC-Patienten leben etwa 10 bis 20 Jahre länger als Menschen mit HbSS.
Sichelzellanämie ist out!
Von „Sichelzellanämie“ sollte man nicht mehr sprechen, denn erstens hat nicht jeder Patient eine Blutarmut und zweitens handelt es sich bei den beschriebenen Hämoglobinopathien um komplexe, lebensverkürzende Erkrankungen, die zum Teil schwere Schäden an ganz verschiedenen Organen anrichten können.
Aber: Patienten mit HbSC-Sichelzellkrankheit haben ein hohes Risiko für Erblindung und Taubheit. Rund 70 % der Erwachsenen mit HbSC erkranken an einer proliferativen Retinopathie, die unbehandelt einen schweren Visusverlust oder Blindheit nach sich ziehen kann. HbSC-Patienten sollten sich daher ab dem siebten Lebensjahr jährlich einer Retinoskopie unterziehen, damit man bei Bedarf frühzeitig mit einer Laser-Koagulation eingreifen kann. Auch der Einsatz einer Anti-VEGF*-Therapie wird heute bei HbSC-Patienten diskutiert.
Manche HbSC-Betroffene weisen hohe Hämoglobinwerte und eine sehr hohe Blutviskosität auf, was Schmerzen, Schwindel oder otologische Probleme nach sich ziehen kann. Wenn es bei ihnen zu einem Hörsturz kommt, sollte sofort ein Aderlass durchgeführt werden, um den Hb-Wert unter 10 g/dl zu senken. Kortison zu geben ist kontraindiziert, weil es die Blutviskosität durch die entstehende Granulozytose noch verstärken und den Hörverlust irreversibel machen könnte. Allgemein brauchen 30–45 der HbSC-Patienten regelmäßig Aderlässe, um schmerzfrei zu bleiben.
* Vascular Endothelial Growth Factor
Quelle: Dickerhoff R. Dtsch Med Wochenschr 2022; 147: 1259-1265; DOI: 10.1055/a-1767-8315