Organmanifestationen bei SSc Systemische Sklerose kennt fast keine Grenzen
 Die systemische Sklerose zeigt sich in einem heterogenen Krankheitsbild - für den Krankheitsverlauf ist eine frühe Diagnose entscheidend.
© Aleksandr – stock.adobe.com
Die systemische Sklerose zeigt sich in einem heterogenen Krankheitsbild - für den Krankheitsverlauf ist eine frühe Diagnose entscheidend.
© Aleksandr – stock.adobe.com
Die systemische Sklerose (SSc) hat eine Vielzahl klinischer Auswirkungen, die die Betroffenen nicht nur in ihrer Lebensqualität erheblich belasten. Sie weist auch unter allen rheumatischen Erkrankungen die höchste Sterblichkeitsrate auf. Die frühe Diagnose der SSc gilt deshalb als essenziell, schreiben Dr. Elizabeth Volkmann von der Universität von Kalifornien in Los Angeles und Kollegen.
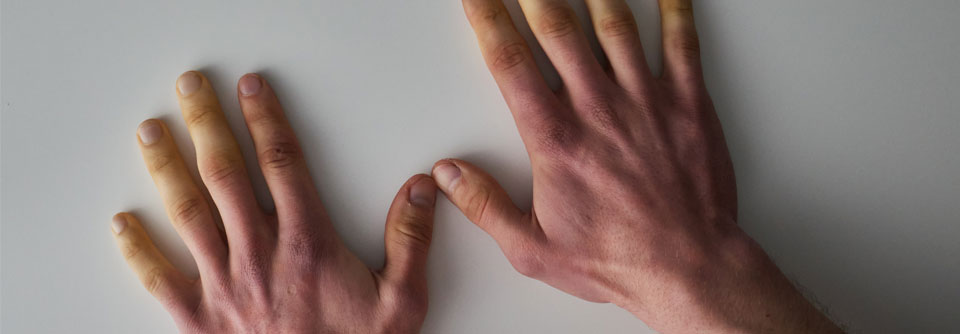
Aufgrund des heterogenen Krankheitsbildes ist das aber kein leichtes Unterfangen. Zu den ersten Anzeichen gehören bekanntermaßen Raynaud-Syndrom und Fatigue. Beide können jedoch mehrere alternative Ursachen haben.
Ob ein Patient mit Raynaud-Phänomen eine SSc entwickelt, lässt sich Untersuchungen zufolge zu einem gewissen Grad vorhersagen. So leiden Patienten mit zusätzlichen sklerodermietypischen Befunden in der Nagelfalz-Kapillarmikroskopie plus spezifischen SSc-Antikörpern nach fünf Jahren zu 65,9 % und nach zehn Jahren zu 72,7 % an einer definitiven SSc. Beim Vorliegen von Raynaud-Symptomatik, positiven SSc-Antikörpern und geschwollenen Fingern waren nach fünf Jahren 94 % an einer SSc erkrankt. Raynaud-Betroffene mit normaler Kapillarmikroskopie ohne SSc-Antikörper litten nach fünf Jahren nur zu 1,3 % an einer SSc.
Bei der Diagnose sowie zur Einschätzung der Progression und des Risikos für Organbeteiligungen helfen Autoantikörper. Bis zu 95 % der Erkrankten sind ANA-positiv, je nach Manifestation zeigen sich vermehrt einzelne spezifischere Antikörper (siehe Tabelle). Eine kleine Subgruppe der Betroffenen weist zwar positive antinukleäre, jedoch keine SSc-spezifischen Antikörper auf. Diesen Patienten drohen eine besonders schnelle Krankheitsprogression und eine damit verbundene höhere Mortalität.
| Spezifische Antikörper bei SSc | |
|---|---|
| Zielstruktur der Antikörper | Klinisches Korrelat |
| Centromer (ACA) | pulmonale Hypertonie |
| Scl-70 | progressive IDL |
| RNA-Polymerase 3 (RNAP III) | renale Krise, Malignität |
| U1-Ribonukleoprotein | Mischkollagenose |
| PM-Scl | Myositis |
Neben den Antikörpern tragen weitere Biomarker zur Krankheitseinschätzung bei:
-
CRP: Hohe Werte sind mit einem erhöhten Risiko für fortschreitende Fibrose, pulmonale Hypertonie und Mortalität assoziiert.
-
Interleukin-6, Krebs-von-den-Lungen-6 and Chemokin-Ligand-18: Hohe Spiegel deuten auf eine interstitielle Lungenerkrankung (ILD) und einen schweren Krankheitsverlauf hin.
-
NTproBNP: Dieses Peptid fungiert als Marker für eine frühe pulmonalen Hypertonie.
Weil SSc-Patienten ein erhöhtes Malignomrisiko aufweisen, sollten sie alle altersentsprechenden Vorsorgeuntersuchungen wahrnehmen. Ein Risikofaktor ist das Vorhandensein von RNA-Polymerase-3-Antikörpern (Anti-RNAP-III). Anti-RNAP-III-positive Patienten sind unabhängig vom Alter auf Malignome zu screenen.
Zukünftig könnte auch die RNA-Sequenzierung an Bedeutung gewinnen. Beispielsweise wurde in einer Studie nachgewiesen, dass bestimmte Transkript-Modulscores das Ansprechen von SSc-Patienten mit ILD auf eine Behandlung mit Mycophenolat vorhersagen.
Haut
Bei der klinischen Beurteilung fällt naturgemäß zunächst die Haut ins Auge. Die kutanen Veränderungen beginnen distal an den Fingern beider Hände und breiten sich nach proximal aus. Es kommt zu Ulzera, Hautverdickungen, Schwellungen, Sklerodaktylie, Kontrakturen und Hautatrophie. Zum Nachweis einer Progression wird meist der modifizierte Rodnan Skin Score (mRSS) herangezogen. Alternativ lässt sich die Hautfibrose über Ultraschall und Durometrie oder zukünftig evtl. der Kombination aus optischer Kohärenzelastografie und optischer Kohärenztomografie bewerten. Auch wenn die allermeisten SSc-Patienten Hautveränderungen aufweisen: Nicht vergessen darf man die kleine Untergruppe von <5 % der Sklerodermiepatienten, die keine Hautfibrosen entwickeln und definitionsgemäß an einer Sklerose ohne Sklerodermie leiden.
Lunge
Eine Lungenbeteiligung gehört zu den entscheidenden Organmanifestationen bei der SSc. Sie tritt bei 50–65 % der Patienten auf. Wenn der Verlauf der ILD auch individuell heterogen ist, so gilt sie doch als häufigste SSc-Todesursache. Risikofaktor für eine progressive ILD sind männliches Geschlecht und das Vorliegen von Scl70-Antikörpern.
Um die Komplikation frühzeitig zu erkennen, sind regelmäßige Lungenfunktionsprüfungen (CO-Diffusionskapazität und forcierte Vitalkapazität) erforderlich. Sensitivste Bildgebung für die SSc-ILD ist die High-Resolution-CT. Sie bietet den Vorteil, dass sie auch andere Manifestationen wie dilatierte Pulmonalarterien und Veränderungen am Ösophagus erkennen lässt. Invasive Maßnahmen wie eine bronchoalveoläre Lavage oder eine Biopsie sind nur angezeigt, wenn die Diagnose unsicher ist.
Niere
Eine weitere Komplikation ist die SSc-bedingte renale Krise. Sie kann unbehandelt innerhalb weniger Wochen zur terminalen Niereninsuffizienz führen. Seit der Einführung der ACE-Hemmer hat sich die Mortalitätsrate aber drastisch reduziert. Wichtigste Maßnahme zur frühzeitigen Diagnose ist die regelmäßige Blutdruckmessung.
Manche Patienten sind allerdings trotz renaler Krise normotensiv, was an deren unzureichender kardiovaskulärer Reserve liegen kann und mit einer schlechten Prognose verbunden ist. Die Nierenkomplikation hat spezifische Risikofaktoren. Dazu gehören Perikarditis, Sehnenreiben/-knirschen, vorherige Prednisolongaben (> 15 mg/d) sowie das Vorliegen von Anti-RNAP-III-Antikörpern. Da die renale Krise meist von einer Thrombozytopenie und einer mikroangiopathisch-hämolytischen Anämie begleitet wird, sind frühzeitige Blutausstriche hilfreich. Nierenbiopsien werden nur in unklaren Fällen empfohlen.
Herz und Gefäße
Die SSc geht mit schweren kardialen Manifestationen einher. Dazu gehören Myokarditis, kongestive Herzinsuffizienz, Herzrhythmusstörungen, asymptomatische fokale Fibrose und gestörte ventrikuläre Relaxation. Sie sind für einen erheblichen Teil der SSc-bedingten Todesfälle verantwortlich. Daher sollten alle SSc-Patienten unabhängig von kardialen Symptomen frühzeitig mit EKG und Echokardiografie untersucht werden. Bei Hinweisen auf eine kardiale Beteiligung folgt eine MRT, drohen maligne Arrhythmien, ist ein Langzeit-EKG erforderlich.
Ernst genommen werden muss auch die pulmonale Hypertonie. Ihre Drei-Jahres-Mortalitätsrate liegt bei 21–48 %. Risikofaktoren dafür sind ausgeprägte Teleangiektasien, eine lange Krankheitsdauer sowie das Vorhandensein von Anti-Zentromer-Antikörpern. Lungenfunktionsprüfungen (vor allem die Messung der isolierten Diffusionskapazität) und Echokardiografie helfen auf dem Weg zur Diagnose. Als Goldstandard zur Diagnose und Ursachenforschung einer pulmonalen Hypertonie gilt der Rechtsherzkatheter.
Magen-Darm-Trakt
Bei 90 % der Patienten ist das Verdauungssystem involviert. Das macht sich häufig klinisch bemerkbar, z.B. durch Dysphagie, Ösophagitis und Refluxbeschwerden bei Ösophagusbeteiligung oder starkem Völlegefühl bei verzögerter Magenentleerung. Auch das GAVE*-Syndrom ist häufig; vor allem anämische Patienten sollten dahingehend endoskopisch untersucht werden. Bei jedem zweiten SSc-Patient ist der untere Gastrointestinaltrakt beteiligt. Mögliche Beschwerden sind Obstipation, Diarrhö, Malabsorption und Stuhlinkontinenz.
Muskeln und Skelett
Muskuloskelettale Manifestationen werden bei der SSc oft übersehen. Gelenkschmerzen sind häufig, und bei manchen Betroffenen kommt es auch zu einer RA. Deren Diagnose ist durch die krankheitsbedingten Hautverdickungen bzw. -schwellungen und Kontrakturen oft schwierig. Generell gelten Gelenkveränderungen (u.a. Gelenkknirschen) bei der SSc als Indiz für eine schlechtere Prognose.
Zu Myopathien verschiedenster Ursachen kann es im Rahmen einer SSc ebenfalls kommen. Die assoziierten Schmerzen und erhöhten CK-Werte verschwinden meist mit Beginn der Immuntherapie. Bei manchen Patienten überlappt die SSc mit einer echten immunvermittelten Myositis. In diesen Fällen steigen die Muskelenzyme deutlich an.
* Gastric antral vascular ectasis
Quelle: Volkmann ER et al. The Lancet, 2022; 401(10373): 304-318, doi.org/10.1016/S0140-6736(22)01692-0.