
Dem plötzlichen Herztod zuvorkommen
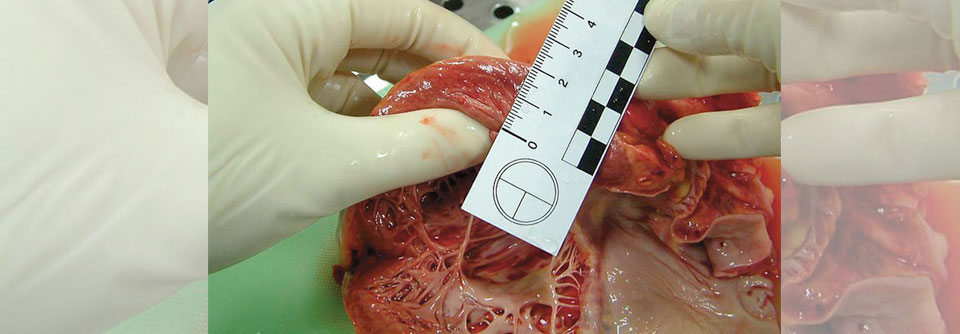 Findet sich keine andere Ursache für die Myokardverdickung, spricht man ab 15 mm von einer hypertrophen Kardiomyopathie.
© wikimedia/Reza luke
Findet sich keine andere Ursache für die Myokardverdickung, spricht man ab 15 mm von einer hypertrophen Kardiomyopathie.
© wikimedia/Reza luke
Die Prävalenz der hypertrophen Kardiomyopathie (HCM) wird weltweit auf einen Erkrankungsfall pro 200 bis 500 Erwachsene geschätzt. Allerdings sind viele Betroffene asymptomatisch und fallen daher klinisch nicht auf, schreiben Steve Ommen von der Mayo Clinic, Rochester, und Christopher Semsarian von der University of Sydney. Bei der HCM liegt eine Verdickung der linksventrikulären Wand vor, für die keine Ursache (wie etwa Hypertonie oder Aortenklappenstenose) verantwortlich gemacht werden kann.
Obwohl die Erkrankung im Allgemeinen als autosomal-dominant vererbte Störung gilt, überwiegen Männer bei den klinisch erfassten Fällen leicht. Diagnostiziert wird die HCM manchmal schon bei der pränatalen Untersuchung. Sie kann sich bei Menschen, die zuvor keine Auffälligkeiten der linksventrikulären Wanddicke aufwiesen, aber auch erst im fünften oder sechsten Lebensjahrzehnt bemerkbar machen.
Erkrankung des Sarkomers
In den meisten Fällen wird die hypertrophe Kardiomyopathie als autosomal-dominantes Merkmal vererbt. Dabei sind verschiedene Mutationen von Genen beschrieben, die für Sarkomerproteine kodieren; diese Proteine spielen für die kontraktile Funktion des Myokards eine wichtige Rolle.
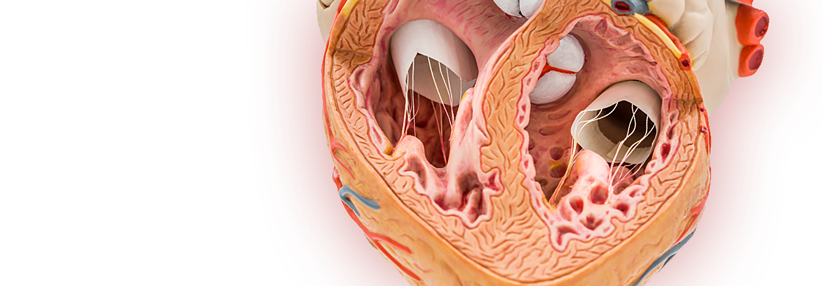
Häufig auffälliger Mitralklappenapparat
Auch wenn die (unerklärte) linksventrikuläre Hypertrophie das wichtigste Merkmal der klinisch manifesten hypertrophen Kardiomyopathie ist, finden sich oft zusätzlich Auffälligkeiten des Mitralklappenapparats. Die veränderten anatomischen Verhältnisse können zudem zu funktionellen Problemen führen, z.B. zu einer Obstruktion der linksventrikulären Ausflussbahn. Eine HCM wird meist in folgenden Situationen entdeckt:- Der Patient berichtet über belastungsabhängige Beschwerden wie Dyspnoe, Angina bzw. Präsynkope/Synkope.
- Es erfolgt ein systematisches Familien-Screening, weil ein Angehöriger von der Erkrankung betroffen ist.
- Bei der Abklärung eines anderen medizinischen Problems fällt die HCM als Zufallsbefund auf.
Herzinsuffizienz entsprechend der Leitlinie behandeln
Ein Teil der HCM-Patienten benötigt spezielle Behandlungsansätze wie die Gabe der stark negativ inotrop wirksamen Substanz Disopyramid oder eine septale Myektomie bzw. eine perkutane Septumablation. Manche Patienten entwickeln auch schwere Herzinsuffizienzsymptome, die dann entsprechend der aktuellen Herzinsuffizienz-Leitlinien behandelt werden müssen.Quelle: Ommen SR, Semsarian C. Lancet 2021; DOI: 10.1016/S0140-6736(21)01205-8
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).
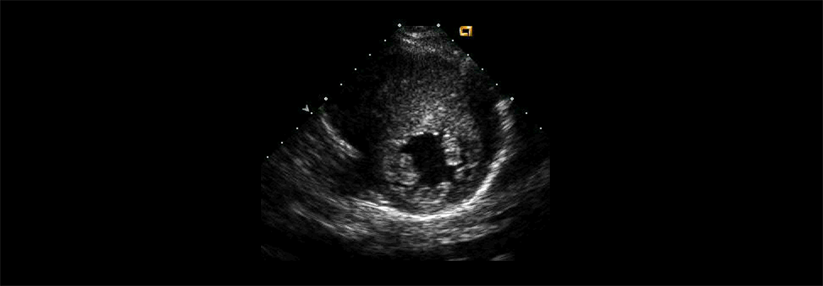

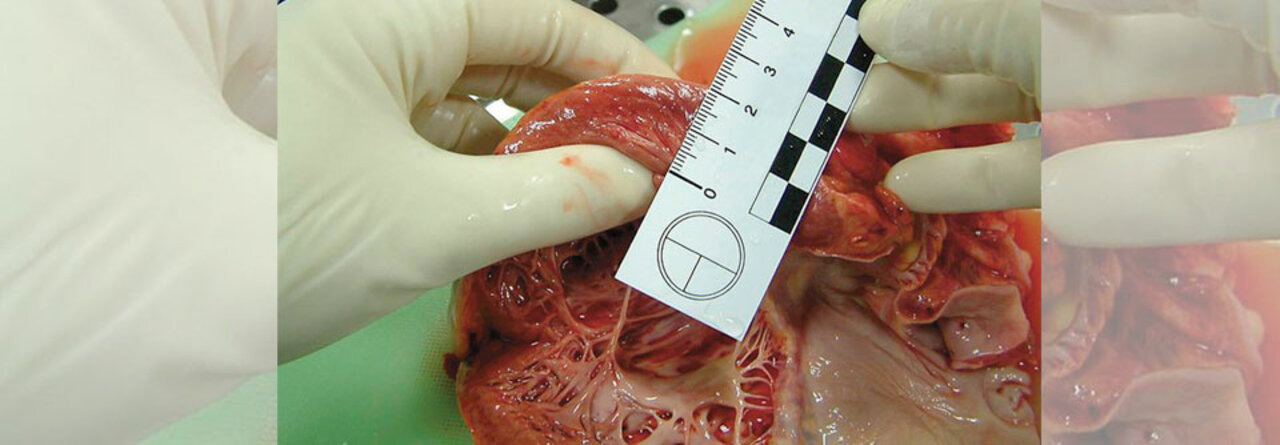 Findet sich keine andere Ursache für die Myokardverdickung, spricht man ab 15 mm von einer hypertrophen Kardiomyopathie.
© wikimedia/Reza luke
Findet sich keine andere Ursache für die Myokardverdickung, spricht man ab 15 mm von einer hypertrophen Kardiomyopathie.
© wikimedia/Reza luke