ILD-Management nur noch im Team?
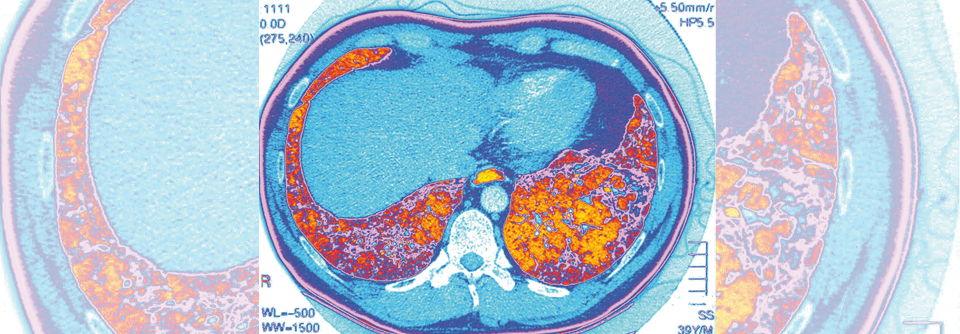
 Die neue S1-Leitlinie zur ILD-Diagnostik wurde im Januar veröffentlicht und enthält u.a. auch einen Patientenfragebogen, der die Anamnese erleichtern soll.
© magicmine – stock.adobe.com
Die neue S1-Leitlinie zur ILD-Diagnostik wurde im Januar veröffentlicht und enthält u.a. auch einen Patientenfragebogen, der die Anamnese erleichtern soll.
© magicmine – stock.adobe.com
Früher konnte man das ILD-Board umgehen, wenn Klinik und HRCT eindeutig für eine interstitielle Lungenfibrose sprachen. Dies hatte zur Folge, dass typische Fälle gar nicht mehr besprochen wurden. Patienten mit eindeutiger IPF wurden also benachteiligt, weil sich kein Expertengremium mit ihrem Fall beschäftigte, erklärte Prof. Dr. Dirk Skowasch, Chef der Pneumologie am Universitätsklinikum Bonn. Heute geht das nicht mehr, denn die neuen Leitlinien sehen vor, dass jeder Patient dem Board vorgestellt wird.
Die Anfang Januar erschienene deutsche S1-Leitlinie zur ILD-Diagnostik1 stellt im Anhang sogar einen Vorschlag für das Protokoll eines solchen Boards vor. Als weitere Schmankerl finden sich ein Konzept für den Arztbrief und ein Patientenfragebogen, den Prof. Skowasch als ausnehmend hilfreich empfiehlt: „Ich drucke ihn jedes Mal aus bei neuen Patienten und lasse die acht Seiten ausfüllen.“ Das gewährleistet, dass bei der Anamnese nichts vergessen wird. Spannend findet Prof. Skowasch die Frage 2.1: Was ist Ihrer Meinung nach die Ursache Ihrer aktuellen Lungenerkrankung? „Da kommt manchmal sogar das Richtige raus.“
Neben der Anamnese gehören Klinik, Funktionsdiagnostik und HRCT zu den Basics, wobei die CT ohne Kontrastmittel erfolgen sollte. Da viele rheumatische Systemerkrankungen mit einer ILD einhergehen können, empfehlen die Leitlinienautoren, eine Autoimmunserologie durchzuführen. Je nach rheumatologischer Verdachtsdiagnose werden u.a. Rheumafaktor, antinukleäre Antikörper, ANCA oder der „Myositis-Blot“ bestimmt. Letzterer umfasst die Messung verschiedener Antikörper, von denn zwölf in der Leitlinie empfohlen werden.
Welche Diagnostik noch erforderlich ist, entscheidet sich nach einer zweiten Board-Konferenz. Meist wird es auf eine Bronchoskopie mit Lavage hinauslaufen, weil dieses wenig invasive Verfahren bei unklarer ILD oft aussagekräftige Resultate liefert. Allerdings können bestimmte HRCT-Muster in Kombination mit klinischen Konstellationen so eindeutig sein, dass die BAL keinen Zusatznutzen bringt. „Wenn ich einen Rheumapatienten habe mit UIP-Muster, ist das eine Rheumalunge – da kann ich auf die Lavage verzichten“, verdeutlichte Prof. Skowasch. Ob noch eine Kryobiopsie erforderlich ist, muss das Board klären. In Bonn diskutiert es in der Regel erst nach erfolgter Lavage, ob eine Biopsie zusätzliche Informationen mit therapeutischen Konsequenzen erwarten lässt.
Wird eine interstitielle Lungenfibrose diagnostiziert, sollte gemäß des 2022-Updates der S2k-Leitlinie „Pharmakotherapie der idiopathischen Lungenfibrose und anderer progredienter pulmonaler Fibrosen“ unverzüglich die antifibrotische Therapie eingeleitet werden.2 Diese Empfehlung stößt zwar nicht auf uneingeschränkte Zustimmung (siehe Kasten), Prof. Skowasch ist jedoch davon überzeugt, dass sich durch die Behandlung akute Exazerbationen, die mit einer äußerst schlechten Prognose einhergehen, abwenden lassen. Er würde nur in Einzelfällen abwarten, ob die IPF progredient verläuft, etwa wenn es sich um einen Zufallsbefund mit geringer Ausprägung handelt oder der Patient zusätzlich z.B. ein Bronchialkarzinom hat. Der Kollege erinnerte daran, welch hohen Benefit die antifibrotische Therapie bringt. Deutsche Registerdaten zeigen nahezu eine Verdopplung der Ein-Jahres-Überlebensrate im Vergleich zu Patienten ohne antifibrotische Behandlung.3 Ähnliche Daten gibt es aus randomisierten, kontrollierten Studien.
Gut informiert mit LeiLa
Die beiden neuen deutschen Leitlinien sind schon LeiLa integriert (www.leila.de). Die App lässt sich auf dem Smartphone oder Tablet, aber auch als Web-Version auf dem PC nutzen. Dazu wird die Identifikationsnummer benötigt, die unter dem Menüpunkt „Einstellungen“ in der App zu finden ist. Die Leitlinien lassen sich mit verschiedenen Funktionen nutzen – als Vollversion, konzentriert auf die Empfehlungen oder per Volltextsuche. Außerdem kann man sich per Mail benachrichtigen lassen, sobald es Neues bei LeiLa gibt.
Von den internationalen Leitlinien hat die deutsche Pharmakotherapieleitlinie2 den Begriff der progredienten pulmonalen Fibrose (PPF) übernommen; er entspricht der „alten“ PF-ILD. Was die PPF-Definition angeht, weicht sie jedoch in einem Punkt ab: Die Lungenfunktionsprüfung gilt den deutschen Experten nicht als conditio sine qua non, um die funktionellen PPF-Kriterien zu erfüllen. Sie sei bei einigen Patienten etwa nach einer akuten Exazerbation gar nicht durchführbar.
Antifibrotisch behandelt werden sollten PPF-Patienten, wenn andere für die jeweilige Diagnose angemessene Behandlungen (z.B. antiinflammatorische Therapie, Expositionskarenz) keine ausreichende Wirkung gezeigt haben – so steht es in der Leitlinie.
Für die Entscheidung – einleiten oder nicht – ist der Befall des Lungenparenchyms ein kritischer Parameter: „Wenn es weniger als 10 % sind, brauchen wir über eine antifibrotische Therapie gar nicht nachzudenken“, so Prof. Skowasch. Zweites wichtiges Kriterium ist die Progression hinsichtlich Klinik, Funktion und/oder Bildgebung. Von einer PPF mit unmittelbarem Therapiebedarf ist auszugehen, wenn die FVC binnen 24 Monaten um 10 % oder mehr abnimmt. Natürlich kann es auch schneller gehen. Hat ein Patient binnen drei Monaten 10 % FVC eingebüßt, muss man nicht 24 Monate warten, um die Behandlung einzuleiten.
Klinik ist nicht Praxis, oder?
Die akute Exazerbation der IPF ist definiert als akute respiratorische Verschlechterung bei bekannter oder aktueller IPF-Diagnose. Sie geht mit neuen bilateralen Milchglasverschattungen und/oder Konsolidierungen einher, die nicht durch kardiale Ursachen oder Flüssigkeitsüberladung erklärbar sind. Die meisten Kollegen geben in solch einem Fall Kortison und Antibiotika, obwohl es dafür keine Evidenz gibt, erklärte Prof. Skowasch. Was man mittlerweile weiß ist, dass sich die Gabe von Cyclophosphamid negativ auswirkt, sie führt zu einer Übersterblichkeit von ca. 10 %, wie eine Studie ergab. Was die Arbeit aber auch zeigte: Eine antifibrotische Vortherapie reduziert das Sterberisiko um zwei Drittel, betonte der Kollege. Insgesamt geht die akute Exazerbation der IPF mit einer extrem schlechten Prognose einher. Innerhalb von drei Monaten stirbt jeder dritte Patient. „Das ist der Grund, weshalb ich jeden IPF-Patienten ab der Diagnose antifibrotisch behandle“, betonte der Bonner Pneumologe.
In der Klinik mag das gerechtfertigt sein, aber Niedergelassene behandeln ganz andere Patienten, widersprach Norbert Mülleneisen, Leverkusen. Er führte als Beispiel eine seiner Patientinnen an, die seit neun Jahren mit einer stabilen IPF lebt: „Da scheue ich mich, ein nebenwirkungsreiches Medikament zu geben oder sie einem Board vorzustellen, in dem zehn Leute sitzen, die sie alle nicht kennen, aber meinen, sie müsse unbedingt behandelt werden.“ Schon recht, entgegnete Prof. Skowasch, aber ihm gehe es nicht um Patienten, die ein niedergelassener Kollege schon seit Jahren stabil führt. „Wenn ein Patient aufschlägt mit frisch diagnostizierter IPF, sollte man die Therapie einleiten, denn Sie wissen nicht, ob er stabil bleibt.“
Von den Antifibrotika ist Nintedanib aktuell das einzige für die PPF zugelassene. Erweist sich die Substanz als unwirksam oder unverträglich, kann – nach Off-Label-Antrag – Pirfenidon versucht werden. Ein neues Wirkprinzip steht mit der PDE-4B-Inhibition ins Haus. Der Wirkstoff mit dem Kürzel BI 1015550 hat in einer Phase-2-Studie an 147 IPF-Patienten mit und ohne antifibrotische Vortherapie vielversprechende Resultate geliefert. Die FVC, die unter Placebo rasant abfiel, nahm unter dem PDE-4B-Hemmer sogar etwas zu und blieb dann über zwölf Wochen nahezu stabil. Die Phase-3-Studien FIBRONEER- IPF und FIBRONEER-ILD sind im Oktober 2022 angelaufen. „Bevor man Nintedanib-Nonresponder auf Pirfenidon umsetzt, kann man auch daran denken, Patienten in eine dieser Studien einzuschließen“, meinte Prof. Skowasch.
Klinische und lungenfunktionelle Verlaufskontrollen sollten alle drei bis sechs Monate erfolgen, wobei das Intervall bei stabilen Patienten auch länger ausfallen darf. Bei Progredienz sollte eine CT veranlasst werden, die anders als zur Diagnosestellung möglichst in Low-dose erfolgen sollte. „Das habe ich bisher nicht gemacht, werde es aber zügig umsetzen – der Verlauf lässt sich auch per Low-Dose-CT beurteilen“, so der Bonner Kollege.
Quelle: Kongressbericht Kongress der Westdeutsche Gesellschaft für Pneumologie (WDGP) 2023
1. S1-Leitlinie Interdisziplinäre Diagnostik interstitieller Lungenerkrankungen im Erwachsenenalter; AWMF Registernr. 020-028;
www.awmf.org
2. S2k-Leitlinie Pharmakotherapie der idiopathischen Lungenfibrose (ein Update) und anderer progredienter pulmonaler Fibrosen; AWMF Registernr. 020-025; www.awmf.org
3. Behr J et al. ERJ 2020; 56: 1902279
4. Richeldi L et a. NEJM 2022; 386: 2178-2187
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).



 Die neue S1-Leitlinie zur ILD-Diagnostik wurde im Januar veröffentlicht und enthält u.a. auch einen Patientenfragebogen, der die Anamnese erleichtern soll.
© magicmine – stock.adobe.com
Die neue S1-Leitlinie zur ILD-Diagnostik wurde im Januar veröffentlicht und enthält u.a. auch einen Patientenfragebogen, der die Anamnese erleichtern soll.
© magicmine – stock.adobe.com