Wenn die Lungenfibrose die Luft abschnürt
 Entzündungsprozesse und Vernarbungen führen bei idiopathischer pulmonaler Fibrose zu respiratorischer Insuffizienz.
© Science Photo Library/Cavallini, James
Entzündungsprozesse und Vernarbungen führen bei idiopathischer pulmonaler Fibrose zu respiratorischer Insuffizienz.
© Science Photo Library/Cavallini, James
Unter interstitiellen Lungenerkrankungen (ILD) versteht man eine Gruppe diffuser parenchymaler Lungenerkrankungen mit unterschiedlichen klinischen, radiologischen und pathologischen Manifestationen. Manche ILD gehen wie die idiopathische Lungenfibrose (IPF) mit einer progredienten Fibrosierung einher – Experten sprechen von progredienter Pulmonalfibrose (PPF; s. Kasten). Die PPF ist charakterisiert durch Abnahme der Lungenfunktion, schlechtes Ansprechen auf immunmodulatorische Therapien und frühe Mortalität.
Wann liegt eine progrediente Pulmonalfibrose vor?
Folgende Kriterien sprechen für eine PPF:
- interstitielle Lungenerkrankung, die nicht einer IPF entspricht
- radiologische Zeichen einer Lungenfibrose
- Evidenz für Progression; hierzu müssen mindestens zwei von drei Kriterien im letzten Jahr erfüllt worden sein, für die sich keine andere Erklärung findet:
- Verschlechterung respiratorischer Symptome
- absolute Abnahme der FVC um > 5 % des Erwartungswerts oder absolute Abnahme der DLCOc* ≥ 10 % des Erwartungswerts innerhalb eines Jahres
- bestimmte radiologische Progressionszeichen
* DLCOc = Diffusionskapazität der Lunge für Kohlenmonoxid, korrigiert für Hämoglobin
IPF und PPF weisen einige gemeinsame Signalwege auf, die – unabhängig vom initialen Trigger – zu einer anhaltenden Fibrosierung führen, schreibt das Team um Gabrielle Liu vom Department of Medicine, Northwestern University, Chicago.
Assoziierte rheumatische Erkrankungen fördern PPF
Allerdings beginnt die PPF oft mit einer inflammatorischen Phase, die durch ein endogenes Autoantigen oder ein exogenes Antigen getriggert wird. Zu den ILD, die am häufigsten zu einer PPF führen, zählen die interstitielle Lungenerkrankung bei rheumatoider Arthritis, systemischer Sklerose, bei Myositis, Sarkoidose u.a. Jedoch variiert der Anteil der Patienten mit diesen ILD, die eine PPF entwickeln, sehr stark.
Nicht nur aus prognostischen Gründen ist es wichtig, Patienten mit PPF zu identifizieren. Denn mit dem Antifibrotikum Nintedanib steht ein Wirkstoff zur Verfügung, der nicht nur bei IPF-, sondern auch bei PPF-Patienten die Verschlechterung der Lungenfunktion verlangsamen kann. Für seinen Einsatz bei PPF gibt es starke Evidenz. So wurden in der randomisierten, placebokontrollierten Phase-3-Studie INBUILD Patienten mit verschiedenen PPFs über 52 Wochen mit Nintedanib oder Placebo behandelt. Primärer Endpunkt war die Abnahme der forcierten Vitalkapazität (FVC). Während die FVC-Abnahme unter Placebo bei 188 ml/Jahr lag, betrug sie unter Nintedanib nur 81 ml/Jahr.
Noch keine abschließenden Ergebnisse zu Pirfenidon
Die Datenlage für den Wirkstoff Pirfenidon bei PPF ist weniger robust. Bisher liegen Ergebnisse aus zwei abgeschlossenen randomisierten, kontrollierten Studien der Phasen 2/2b vor. Durch Pirfenidon konnte die Abnahme der Lungenfunktion (gemessen als FVC) in einem Zeitraum von 24 bzw. 48 Wochen gegenüber Placebo verlangsamt werden. Eine der beiden Studien wurde aufgrund der nur zögerlichen Rekrutierung von Teilnehmern vorzeitig beendet. Dennoch könnten die Ergebnisse klinisch relevant sein. Derzeit laufen weitere klinische Studien mit Pirfenidon und Nintedanib bei verschiedenen PPF-Subgruppen.
Welche Rolle spielt die Immunsuppression? Wenn eine immunsuppressive Behandlung bei entzündlichen ILD wirksam ist, wird sie fortgesetzt. Falls es trotz Immunsuppression bei einer inflammatorischen ILD zu einer progredienten Fibrose kommt, stellt sich die Frage, ob die Immunsuppression eskaliert oder eine Therapie mit einem Antifibrotikum wie Nintedanib eingeleitet werden soll. Die Autoren raten, bei der Therapieentscheidung die Zeit seit Krankheitsbeginn mit zu bedenken, da immunsuppressive Therapien im frühen Krankheitsverlauf wahrscheinlich effektiver sind.
Zu den nicht-medikamentösen Behandlungsoptionen zählt die Sauerstofflangzeittherapie. Sie wird bei Patienten mit Ruhehypoxämie empfohlen. Zur Unterstützung von Atmung und Besserung der Lebensqualität ist auch die Lungenrehabilitation nützlich. Ultima Ratio der nicht-medikamentösen Optionen bleibt die Lungentransplantation.
Insgesamt sind im Management der PPF noch viele Wünsche offen, schreiben die Autoren. Aktuell wird intensiv nach Biomarkern gesucht, die frühzeitig eine Vorhersage erlauben, welche ILD-Patienten eine PPF entwickeln werden. Gesucht werden zudem Wirkstoffe, die die pulmonale Fibrosierung stoppen oder sogar heilen können. Im Tierexperiment erwies sich das Antidiabetikum Metformin kürzlich als hoffnungsvolle Substanz: Metformin konnte bei Mäusen eine durch Bleomycin induzierte Lungenfibrose verbessern.
Quelle: Liu GY et al. BMJ 2022; 377: e066354; DOI: 10.1136/bmj-2021-066354
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).


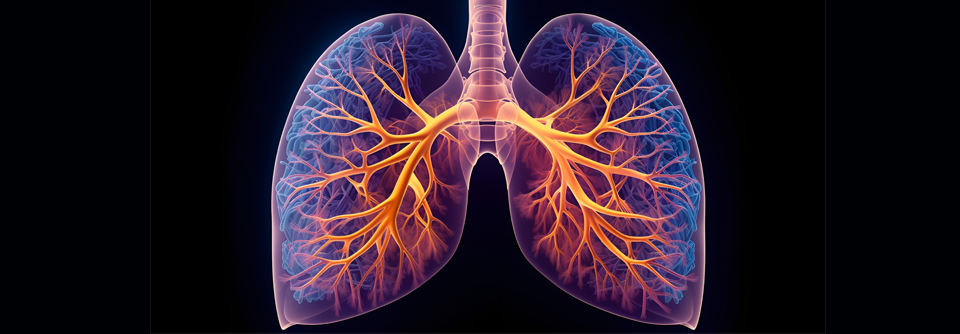

 Entzündungsprozesse und Vernarbungen führen bei idiopathischer pulmonaler Fibrose zu respiratorischer Insuffizienz.
© Science Photo Library/Cavallini, James
Entzündungsprozesse und Vernarbungen führen bei idiopathischer pulmonaler Fibrose zu respiratorischer Insuffizienz.
© Science Photo Library/Cavallini, James