
Kleingefäßvaskulitis ganz groß
 Morbus Behςet kann sich auch am Auge manifestieren.
© Vladimir Voronin – stock.adobe.com
Morbus Behςet kann sich auch am Auge manifestieren.
© Vladimir Voronin – stock.adobe.com
Der Morbus Behçet kann als systemische Vaskulitis Venen und Arterien jeden Kalibers befallen. Seine Pathogenese ist immer noch nicht genau geklärt. Prinzipiell geht man davon aus, dass bei Menschen mit genetischer Prädisposition diverse externe Trigger spezifische Vorgänge anstoßen, die letztendlich in Inflammation und Zellschädigung münden. Als mögliche Triggerfaktoren gelten Mikroorganismen und ihre Stoffwechselprodukte ebenso wie histaminfreisetzende Nahrungsmittel (z.B. Zitrusfrüchte, Nüsse oder Käse). Eine schlechte Mundhygiene und Stress sollen zusätzlich begünstigend wirken, schreiben Dr. David Saadoun von der Sorbonne Universität in Paris und Kollegen. Mehr und mehr kristallisiert sich heraus, dass Disbalancen in der Darm- und Speichelflora bei der Entwicklung der Erkrankung eine Bedeutung haben.
Von der genetischen Komponente wird schon länger ausgegangen, u.a. aufgrund der familiären Häufung in bestimmten Populationen. Dies soll vor allem für mit früh einsetzendem Behçet-Syndrom gelten. Wie sich inzwischen herausstellte, haben Träger der HLA-Antigenvariante HLA-B51 eine sechsfach erhöhte Wahrscheinlichkeit, einen Morbus Behçet zu entwickeln. Studien zufolge gibt es Interaktionen zwischen HLA-B51 und dem Gen, das die Aminopeptidase 1 des endoplasmatischen Retikulums (ERAP1) steuert. Das könnte sich auf die Aktivierung der natürlichen Killerzellen und die T-Zell-Homöostase auswirken. Zahlreiche weitere Gene zur Regulation von Th-1- und Th-17-Zellen oder zur Steuerung der Zellchemotaxis wurden ebenfalls mit der Erkrankung in Verbindung gebracht.
Ähnlich vielfältig wie die pathogenetischen Faktoren sind die klinischen Merkmale des Morbus Behçet. Die Manifestationen überlappen oft, wobei die individuellen Risiken je nach ethnischer Gruppe höher oder niedriger liegen können.
Haut und Schleimhaut
Mit einer Häufigkeit von 98 % entwickeln so gut wie alle Behçetpatienten rezidivierende orale Ulzera. Diese sind von Aphthen anderer Genese oft kaum zu unterscheiden. Bis zu zwei Drittel der Patienten leiden unter genitalen Ulzera, vor allem an den Labien und am Skrotum. Sie sind größer und tiefer als die oralen Läsionen und heilen langsamer und i.d.R. oft narbig ab.
Papulopustuläre, akneiforme Hautmanifestationen gehören ebenfalls zum Erscheinungsbild des Morbus Behçet. Auch noduläre Läsionen kommen vor. Sie können zudem auf eine oberflächliche Thrombophlebitis hindeuten, als Pannikulitis ähneln sie einem Erythema nodosum. Kutane Beschwerden sind beim Behçet nicht nur häufig – bei etwa einem Drittel der Betroffenen bleibt es bei der kutanen Manifestation, schreiben die Autoren.
Gelenke
Etwa die Hälfte der Behçetpatienten entwickelt Gelenkmanifestationen. Meist handelt es sich um nicht-destruierende, selbstlimitierende Arthralgien, insbesondere an Knien, Knöcheln, Handgelenk und Ellenbogen. Manchmal sind begleitend auch Bänder und Sehnen betroffen. Eher ungewöhnlich ist ein Befall der Wirbelsäule und Iliosakralgelenke.
Augen
In 50 % der Fälle kommt es zu einer Augenbeteiligung, bei über drei Viertel der Patienten beidseits und bei den meisten etwa zwei Jahre nach den ersten Behçet-Symptomen. Am häufigsten ist die Panuveitis, weitere Manifestationen sind Glaskörperentzündungen, fokalen Retinitiden oder retinale Gefäßverschlüsse.
Gefäße
Die häufig rezidivierenden Gefäßentzündungen treffen beim Behçet Venen und Arterien gleichermaßen. Mit einer Häufigkeit von bis zu 40 % sind oberflächliche Thrombophlebitis und tiefe Venenthrombose die Spitzenreiter. Letztere kommt vor allem an den unteren Extremitäten vor und zieht oft ein postthrombotisches Syndrom nach sich. Lungenembolien sind allerdings selten.
Im arteriellen System manifestiert sich die Erkrankung sowohl mit Aneurysmata als auch mit Stenosen oder thrombotischen Verschlüssen, insbesondere an der Aorta und den peripheren Arterien. Der Befall der Pulmonalarterien ist zwar selten, aber gilt als sehr spezifisch für den Behçet. Etwa 5 % der Patienten entwickeln eine Herzbeteiligung, z.B. in Form von Perikarditis, Myokarditis, Koronararteriitis, Klappenentzündung oder intrakardialen Thrombosen.
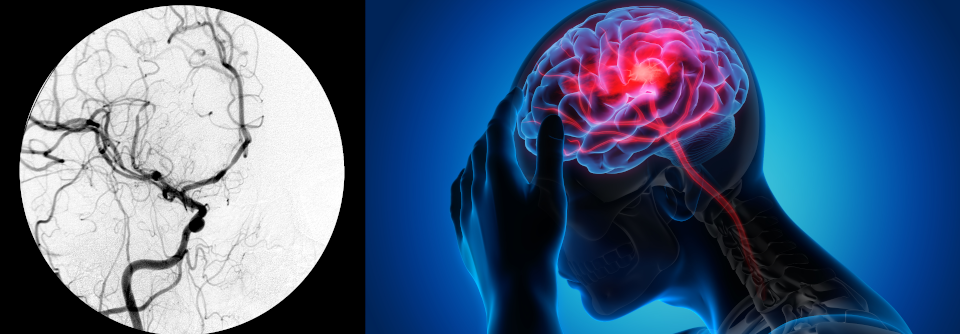
Neurologische Manifestationen
Zu neurologischen Manifestationen kommt es bei rund 10–30 % der Betroffenen. Sie umfassen Entzündungen von Hirnstamm und Mesodienzephalon, Meningitis, Enzephalitis und Myelitis. Mögliche Folgen sind Kopfschmerzen, Hemiparesen und Krämpfe sowie neurokognitive Dysfunktionen. Etwa 25 % der neurologischen Manifestation sind nicht-parenchymatös, wobei sich es in diesen Fällen meist um zerebrale Venenthrombosen handelt, was eine Überlappung zu den Gefäßmanifestationen hinsichtlich der tiefen Venenthrombose darstellt.
Gastrointestinum
Während in Ostasien der Morbus Behçet bei jedem fünften Patienten auch den Gastrointestinaltrakt befällt, ist diese Manifestation in Europa sehr selten (< 5 %). Es drohen Schleimhautulzera, die Schmerzen, Durchfall und schwere Blutungen auslösen können. Die Abgrenzung zu chronisch-entzündlichen Darmerkrankungen ist aufgrund der Beschwerden bei diesen Patienten schwierig.
Unabhängig von der Art der Manifestation ist das Therapieziel beim Morbus Behçet die Entzündungskontrolle und damit das Verhindern irreversibler Organschäden, schreiben Dr. Saadoun und Kollegen. Als First-Line-Behandlung für Haut-, Schleimhaut und Gelenkmanifestationen gilt Colchicin. Bei rezidivierenden Beschwerden werden zusätzlich oft Glukokortikoide eingesetzt, topisch und intraartikulär. Gegen orale Ulzera helfen antientzündliche Mundspülungen und in refraktären Fällen ist Apremilast eine Option. Auch Azathioprin, Thalidomid, Interferon-alpha und Etanercept haben in Studien positive Effekte bei Ulzera gezeigt. Vielversprechend für Betroffene, die nicht auf Colchicin ansprechen, scheint zudem Ustekinumab zu sein.
Hat der Patient bereits Organmanifestationen entwickelt, stehen Glukokortikoide und systemische Immunsuppressiva an erster Stelle. Bei visusbedrohender Uveitis raten die Experten z.B. zu einem Therapieregime, das Infliximab oder IFN-a mit einbezieht.
Adalimumab wurde zumindest von der FDA bei nicht-infektiöser Uveitis zugelassen. Tiefe Venenthrombosen werden mit Glukokortikoiden und konventionellen Immunsuppressiva oder Biologika behandelt. Ob eine Antikoagulation erforderlich ist, wird diskutiert. Arterielle Läsionen erfordern hohe Glukokortikoiddosen, kombiniert mit Azathioprin, Cyclophosphamid oder einem TNF-Inhibitor. Die Maßnahmen senken zudem das Risiko von Komplikationen nach den Revaskularisierungsmaßnahmen. Hoch dosierte Glukokortikoide plus Azathioprin oder Cyclophosphamid werden auch für akute neuroparenchymale Manifestationen empfohlen – hinsichtlich Anti-TNF gibt es nur einzelne, jedoch positive Fallstudien. Patienten mit gastrointestinalem Befall erhalten Glukokortikoide, bei milden Formen kombiniert mit 5-Aminosalicylsäure, bei schwereren mit Azathioprin. In therapierefraktären Fällen lässt sich auf TNF-Hemmer oder Thalidomid ausweichen.
Vor allem für die Therapie der schweren und lebensbedrohlichen Organmanifestationen gibt es bisher keine gute Evidenz, bedauern die Autoren. Meist muss die Wahl zwischen Cyclophosphamid und einem TNF-a-Blocker getroffen werden. Als mögliche neue Option für schwere Fälle gilt Tocilizumab.
Die Prognose variiert nach dem Ausmaß der Erkrankung. Entscheidend ist der Organbefall, wie die Experten schreiben. Junge Männer haben häufiger schwere Verläufe. Insgesamt ist männliches Geschlecht mit einem erhöhten Mortalitätsrisiko assoziiert (Hazard Ratio, HR, 4,94). Auch die Beteiligung von Arterien sowie häufige Krankheitsschübe erhöhen das Sterberisiko (HR 2,51 bzw. 2,37). Als schwerwiegendste Komplikation gilt der Visusverlust. Vor dem Einsatz von Immunsuppressiva waren 25 % der Patienten innerhalb von zehn Jahren nach der Behçet-Diagnose davon betroffen, heutzutage sind es etwa 13 %. Bei einem neurologischen Befall liegt das Risiko für eine schwere Behinderung oder Tod bei 25 % nach sieben und bei 60 % nach zehn Jahren.
Das Vorkommen des Morbus Behςet variiert stark je nach geografischer Region und Ethnie der Bevölkerung. Historisch gesehen trat die Erkrankung vor allem entlang der Seidenstraße auf. Heute findet sich die höchste Prävalenz mit 420 Fällen pro 100.000 Einwohnern in der Türkei. In Europa hat sich ein Nord-Süd-Gefälle herausgebildet, wobei die Prävalenzen von 0,3–4,9/100.000 in den nördlichen und 1,5–15,9/100.000 in den südlichen Ländern reichen. In den Niederlanden und Deutschland war die Häufigkeit des Morbus Behςet bei Immigranten aus Hochprävalenzregionen höher als bei nicht immigrierten Einwohnern, obwohl die Raten bei Ersteren niedriger sind als in ihren jeweiligen Herkunftsländern.
Quelle: Aus der Fachliteratur
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).
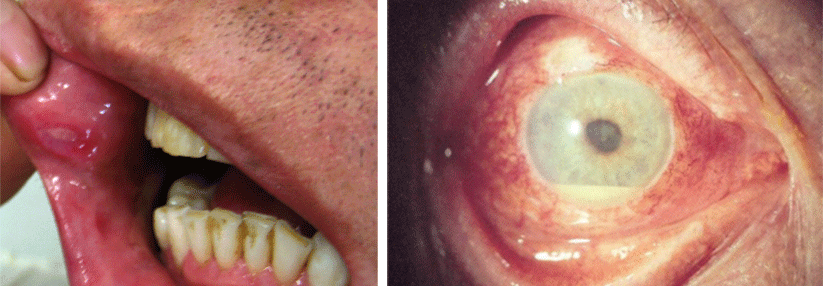


 Morbus Behςet kann sich auch am Auge manifestieren.
© Vladimir Voronin – stock.adobe.com
Morbus Behςet kann sich auch am Auge manifestieren.
© Vladimir Voronin – stock.adobe.com