
Neuromuskuläre Erkrankungen mit Erbgutmanipulation erfolgreich behandeln
 Das Gentherapeutikum Onasemnogen-Abeparvovec gilt als das teuerste Medikament der Welt.
© iStock/metamorworks
Das Gentherapeutikum Onasemnogen-Abeparvovec gilt als das teuerste Medikament der Welt.
© iStock/metamorworks
Die europäische Arzneimittelbehörde EMA definiert Gentherapie als biologisches Medizinprodukt, das mit einer rekombinanten Nukleinsäure eine Gensequenz regulieren, reparieren, ersetzen, hinzufügen oder stilllegen soll. Aus ethischen Gründen ist derzeit lediglich die somatische Gentherapie gestattet, bei der keine veränderte Erbinformation an die nächste Generation weitergegeben wird, nicht aber die auf Keimbahnebene. Das genetische Material kann per viralem Vektor – meist Adeno-assoziierte Viren (AAV) – oder auch per Plasmid mit und ohne liposomale Hülle in die Zielzellen eingeschleust werden.
Mit Schnäppchen ist nicht zu rechnen
Die erste neuromuskuläre Erkrankung, für die eine Gentherapie zur Verfügung steht, ist die spinale Muskelatrophie. Die Zulassung von Onasemnogen-Abeparvovec, besser bekannt unter dem Handelsnamen Zolgensma®, hat für Wirbel gesorgt. Mit mehr als einer Million Euro für eine Spritze gilt es als das teuerste Medikament der Welt. Auch künftige Gentherapien werden wohl keine Schnäppchen sein. Wichtig ist daher, direkte und indirekte Langzeitkosten der unbehandelten Krankheit mit den Therapiekosten ins Verhältnis zu setzen, um Patienten, ihren Angehörigen und der Gesellschaft gleichermaßen gerecht zu werden, meinte Professor Dr. Giuseppe Vita, Universität Messina.
Heute sind rund 200 Produkte für neuromuskuläre Erkrankungen in den therapeutischen Pipelines, berichtete Dr. Teresinha Evangelista vom Hôpital Pitié Salpêtrière der Sorbonne Université, Paris. Darunter befinden sich zu 43 % small molecules, zu 14 % Gentherapien und zu 9 % Antisense-Oligonukleotide, welche die Genexpression modulieren und Gene gezielt ausschalten können. Die Hälfte ist bereits in klinischer Erprobung, Zielerkrankungen sind vor allem die Amyotrophe Lateralsklerose und die Muskeldystrophie Duchenne.
Ein gutes Ziel für die Gentherapie gibt die Charcot-Marie-Tooth-Erkrankung (CMT) ab. Sie ist die häufigste hereditäre Neuropathie mit zwei Haupt- und vielen Unterformen. Sie entsteht monogenetisch, die Zielzellen sind bekannt und die genetischen Mechanismen relativ gut definiert. Für die CMT-Unterform Giant Axon Neuropathie 1 (GAN1), klinisch gekennzeichnet durch rasch progrediente Muskelschwäche und frühen Tod, wurden inzwischen über 40 funktionell wirksame Mutationen im Gigaxonin-Gen identifiziert. Heterozygote Träger solcher Mutationen erkranken nicht, was hoffen lässt, dass eine verstärkte Expression exogener GAN1-Wildtyp-Gene therapeutisch nützlich sein wird, so Dr. Evangelista. Proof-of-concept-Versuche mit einer AAV-basierten Genersatztherapie am GAN1-Mausmodell und an Patienten verliefen erfolgreich. Derzeit gibt es erste klinische Studien mit dem Gentherapeutikum scAAV9/JeT-GAN.
Auch bei monogenetischen kongenitalen Myopathien wie der X-chromosomalen myotubulären Myopathie (XLMTM), einer seltenen Erkrankung, die ausschließlich Jungen befällt, steht man offenbar vor dem Durchbruch. Das erste Kind mit XLMTM wurde bereits 2017 in Boston mit dem AAV8-basierten Gentherapeutikum AT132 behandelt. Derzeit läuft die klinische Prüfung in einer Studie namens ASPIRO. „Die ersten Ergebnisse der Studie sind extrem vielversprechend“, konstatierte Dr. Evangelista.
Im Kommen sind ferner RNA-basierte Therapien, die über mehrere Pfade zum Ziel führen können. Ein Beispiel ist die Transthyretin (TTR)-Amyloid-Neuropathie, eine hereditäre periphere und/oder autonome neuronale Erkrankung. Über 130 Mutationen im TTR-Gen sind bekannt, die zur Fehlfaltung, Instabilität des TTR-Tetramers und Aggregation des in der Leber gebildeten TTR-Proteins führen. Zielorgan für die Behandlung ist hier also nicht die Nervenzelle, sondern die Leber.
Gute Proteinexpression in LGMD-Studie
Zwei Techniken kommen zum Einsatz: ein Antisense-Oligonukleotid namens Inotersen und eine in einem Lipidnanopartikel verpackte small interfering RNA (siRNA) namens Patisiran. Beide reduzieren TTR in Blut und Geweben und dadurch die Progression der Neuropathie. Sie sind seit 2018 zugelassen.
siRNA-Therapien sind auch für die Gliedergürtelmuskeldystrophie (Limb Girdle Muscular Dystrophy, LGMD) in klinischer Erprobung, ebenso fünf „echte“ Gentherapien. Exemplarisch berichtete Dr. Evangelista über eine Phase-1-Studie, in der sechs Patienten mit LMGD2 mit dem per AAV-Vektor verabreichten Gen MYO-102 behandelt werden. Die Laufzeit beträgt zwei Jahre. Erste Resultate zeigen eine gute Proteinexpression in den Muskeln, in die das Gentherapeutikum infundiert wurde.
Für die spinale Muskelatrophie steht schon seit 2016 das erste RNA-basierte Medikament zur Verfügung, berichtete Prof. Vita. Hintergrund ist, dass zwei Gene für dasselbe Protein kodieren. SMN1 erzeugt zu 100 % funktionstüchtiges Survival-Motor-Neuron(SMN)-Protein, SMN2 eine verkürzte und vermindert wirksame Proteinvariante. Bei der SMA ist das SMN1-Gen mutiert oder deletiert, was das Überleben der Motoneuronen drastisch reduziert.
Das Antisense-Oligonukleotid Nusinersen sorgt dafür, dass die Transkription des SMN2-Gens vermehrt funktionstüchtiges Protein erzeugt. Real-World-Erfahrungen zeigen, dass Kinder mit SMA sich unter dem Therapeutikum besser entwickeln, oft keine Beatmung brauchen und länger überleben. Im Unterschied dazu ersetzt die Gentherapie mit Onasemnogen-Abeparvovec das defekte SMN1-Gen. Nusinersen wird nach dem Aufdosieren alle vier Monate verabreicht, bei Onasemnogen-Abeparvovec ist noch nicht absehbar, ob die Einmalapplikation lebenslang ausreicht. Bisher jedenfalls hält die Wirkung fünf Jahre unvermindert an.
Gentherapien sind wohl meist irreversibel
Natürlich bleiben Hürden zu überwinden. So gibt es für viele der seltenen Erkrankungen (noch) keine validierten, spezifischen und sensitiven Messinstrumente, mit denen man den klinischen Erfolg der Therapien messen könnte. Die Beobachtungsstudien, die sie liefern sollen, laufen gerade an. Den Erfolg zu messen, ist angesichts der langsamen klinischen Progredienz mancher Erkrankungen schwierig.
Außerdem steht zu vermuten, dass die Gentherapien umso effektiver wirken, je früher sie im Krankheitsverlauf eingesetzt werden. Asymptomatische Vorstadien zu behandeln, entspricht derzeit aber nicht dem medizinischen Standard. Zudem sind Gentherapien wahrscheinlich meist irreversibel, gab Dr. Evangelista zu bedenken – das gelte für günstige wie für ungünstige Effekte.
Kongressbericht: 6th Congress of the European Academy of Neurology | Onlineveranstaltung
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).
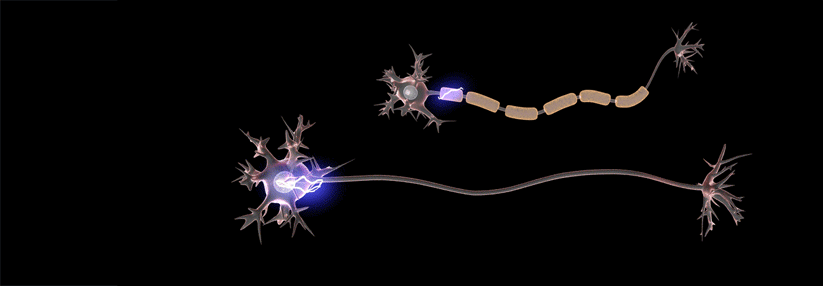


 Das Gentherapeutikum Onasemnogen-Abeparvovec gilt als das teuerste Medikament der Welt.
© iStock/metamorworks
Das Gentherapeutikum Onasemnogen-Abeparvovec gilt als das teuerste Medikament der Welt.
© iStock/metamorworks