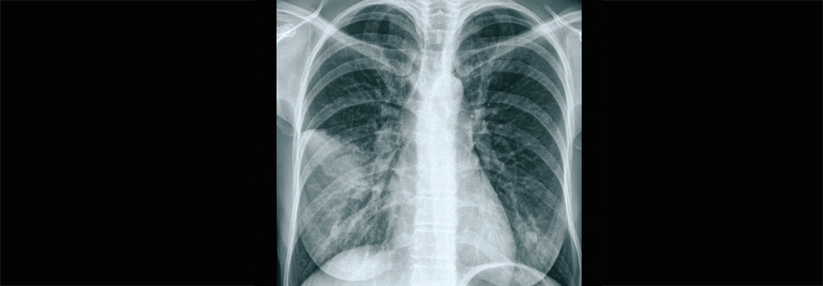
Spezielle Therapie hilft bei der seltenen Alveolarproteinose
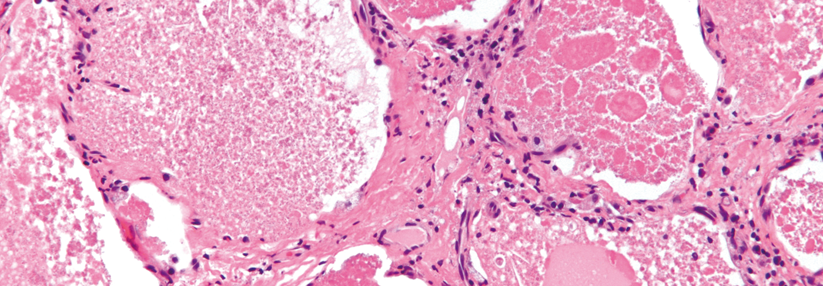 Intraalveolär findet sich ein amorphes eosinophiles feingranuläres Material mit reichlich Lipiden.
© wikimedia commons/Nephron
Intraalveolär findet sich ein amorphes eosinophiles feingranuläres Material mit reichlich Lipiden.
© wikimedia commons/Nephron
Bei der pulmonalen Alveolarproteinose (PAP) sammeln sich Surfactant-Lipoproteine in den Lungenbläschen an. Das behindert den Gasaustausch unterschiedlich stark und führt zu einem breiten Spektrum klinischer Symptome von Belastungsintoleranz bis zu hypoxämiebedingtem Atemversagen.
Die Funktionen des Surfactant
Surfactant ist ein Lipoproteinkomplex, der von Alveolarepithelzellen Typ II in den Alveolarraum sezerniert wird und dort eine membranartige Schicht bildet. Diese übernimmt wichtige Aufgaben: Sie stabilisiert die Lungenvolumina während des Atemzyklus, indem sie die Oberflächenspannung an der Luft-Flüssigkeit-Schnittstelle reduziert. Damit wird verhindert, dass die Alveolen endexspiratorisch kollabieren und sich Atelektasen bilden. Surfactant stabilisiert auch die Alveolargröße, verringert die elastische Rückstellung der Lunge und beteiligt sich an der Abwehr gegen mikrobielle Pathogene. Der Abbau des Surfactant erfolgt durch Alveolarmakrophagen. GM-CSF aktiviert Signalwege, die zur Ausdifferenzierung dieser Alveolarmakrophagen führen.
Es gibt primäre, sekundäre und kongenitale Formen der Erkrankung, schreiben Dr. Elena Salvaterra und Dr. Ilaria Campo von der Abteilung für Innere Medizin der Universität Pavia. Der primären Form liegt eine Störung der GM-CSF*- Signaltransduktion zugrunde, die eine mangelnde Surfactant-Clearance zur Folge hat. Sie kann hereditärer oder autoimmuner Natur sein. Die autoimmune Form zeichnet für 90 % aller PAP-Fälle verantwortlich und beruht auf einer Bildung von anti-GM-CSF-Autoantikörpern. Die hereditäre primäre PAP wird verursacht durch Mutationen des Gens, das für den GM-CSF-Rezeptor kodiert.
Eine sekundäre Alveolarproteinose findet man im Zuge einiger Erkrankungen, die sich u.U. negativ auf die Zahl und/oder Funktion von Alveolarmakrophagen auswirken. Dies sind vor allem hämatologische und chronisch entzündliche Erkrankungen. Auch die Exposition gegenüber Immunsuppressiva oder toxischen Substanzen zählt zu den möglichen Ursachen. Mutationen in Genen, die für Surfactant-Proteine kodieren, verursachen eine kongenitale PAP. Das gebildete dysfunktionale Surfactant kann in diesem Falle seine physiologische Funktion nicht erfüllen.
Nach US-amerikanischen und japanischen Erhebungen hat die PAP eine Prävalenz von etwa 7 pro 1 Million Einwohner. Das mittlere Alter der Patienten zum Zeitpunkt der Diagnose liegt bei etwa 50 Jahren, zu zwei Dritteln trifft es Männer.
Kongentiale PAP kann für Neugeborene tödlich sein
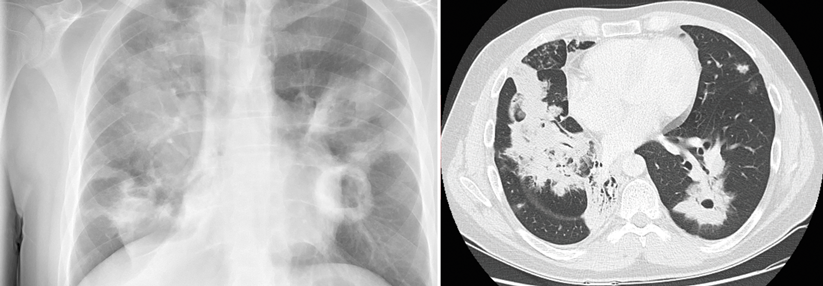
Ein Drittel der Erwachsenen mit autoimmuner Alveolarproteinose ist asymptomatisch. Die übrigen klagen anfangs über Belastungsdyspnoe und eventuell weitere respiratorische Symptome wie Husten und Sputumproduktion. Manche zeigen systemische Symptome, etwa Fatigue und/oder Gewichtsverlust. Die hereditäre Form äußert sich ähnlich, nur bereits in der Kindheit. Dyspnoe und Husten fallen auch bei sekundären Formen auf – im Kontext mit dem Grundleiden. Die kongenitale PAP führt abhängig von der zugrunde liegenden Mutation in einigen Fällen schon in den ersten Lebenstagen zum Tod, bestenfalls bleibt sie bis ins Erwachsenenalter unbemerkt. Fieber kann als eigenständiges Symptom einer Alveolarproteinose vorkommen, was den Verdacht auf eine Pneumonie nahelegt. Radiologische Befunde mit symmetrischen perihilär betonten Infiltraten weisen in die gleiche Richtung, evtl. erfolgen nicht-indizierte Antibiotikatherapien. Die richtige Diagnosestellung verzögert sich. Man braucht daher zwingend eine hrCT. Sie zeigt ein typisches Muster mit interlobulärer Septenverdickung und pflasterartig angeordneten milchglasartigen Verdichtungen („crazy paving“). Spezifisch für die PAP ist dieses Muster allerdings nicht. Aber das Zusammenspiel mit langsam progredienten unspezifischen respiratorischen Symptomen sollte an eine PAP denken lassen. Die Lungenfunktionsprüfung gibt anfangs nicht viel her, erst bei schwerer Ausprägung finden sich Zeichen einer restriktiven Störung. Das Routinelabor hilft ebenfalls kaum weiter, lediglich der Laktatdehydrogenasespiegel steigt oft an. Zur definitiven Diagnose führt die flexible Bronchoskopie mit bronchoalveolärer Lavage. Sie bringt eine undurchsichtige und milchige Flüssigkeit ans Licht, die oft große Mengen an Sediment enthält. Nach der Diagnose folgt die Klassifizierung der PAP. Wegen der weiten Verbreitung der autoimmunen Form sollten zunächst GM-CSF-Autoantikörper im Serum bestimmt werden. Als belegend gelten Titer von mehr als 5 µg/ml-1. Allerdings bieten nur wenige Labore weltweit diese Untersuchung an. Bei normalen Antikörper-Spiegeln bestimmt man die GM-CSF-Konzentration im Serum. Patienten mit hereditären Formen weisen Werte > 10 pg/ml-1 auf. Der Verdacht auf eine kongenitale Alveolarproteinose lässt sich durch Mutationsanalyse der für die Surfactantproduktion relevanten Gene abklären. Zur erweiterten Diagnostik gehört natürlich auch, mögliche Ursachen für eine sekundäre Form zu identifizieren. Die Therapie hängt von der Krankheitsform und ihrem Schweregrad ab und reicht von „watchful waiting“ bei asymptomatischer Erkrankung bis hin zur Ganzlungenlavage. Sie ist derzeit der Goldstandard bei Patienten mit primärer PAP, und manchmal bei sekundärer. Es handelt sich um eine invasive, nicht-standardisierte Prozedur, die nur in spezialisierten Zentren auf der Intensivstation in Allgemeinnarkose Anwendung findet. Der Patient liegt auf der Seite, durch die oben gelegene Lunge fließt warme Kochsalzlösung, während die untere selektiv über einen doppellumigen endobronchialen Tubus ventiliert wird. Die Lavage wiederholt man, bis klare Flüssigkeit kommt. Nach einem festgelegten Intervall erfolgt die Behandlung der anderen Seite. Zu einer Alternative könnte sich die subkutane, besser noch die inhalative Therapie mit GM-CSF entwickeln. In Studien hat sie Lungenfunktion, Belastbarkeit und Schweregrad-Score nicht weniger verbessert als die Lavage.Genetisch korrigierte Makrophagen transplantieren
Als mögliches neues Target wird derzeit Cholesterin erforscht, das sich in den Alveolarmakrophagen von PAP-Kranken überreichlich findet. Einzelne Patienten haben bereits auf Statine gut angesprochen. Bei hereditärer PAP scheint die Transplantation genetisch korrigierter Pulmonalmakrophagen als ein vielversprechender Ansatz.* Granulozyten-Monozyten-Kolonien-stimulierender Faktor
Quelle: Salvaterra E, Campo I. Breathe 2020; 16: 200018; DOI: 10.1183/20734735.0018-2020
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).

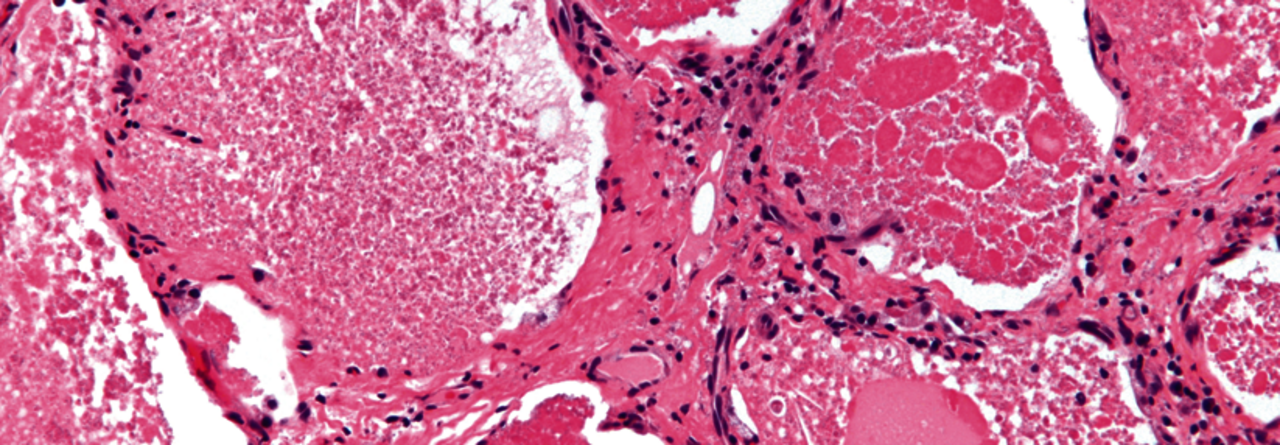 Intraalveolär findet sich ein amorphes eosinophiles feingranuläres Material mit reichlich Lipiden.
© wikimedia commons/Nephron
Intraalveolär findet sich ein amorphes eosinophiles feingranuläres Material mit reichlich Lipiden.
© wikimedia commons/Nephron