Misslungenes Eiweiß-Origami Was man über Amyloidosen wissen sollte
 Letzten Endes führten die Exzisionsbiopsien schließlich zur Diagnose einer AL-Amyloidose.
© Juan Gärtner – stock.adobe.com
Letzten Endes führten die Exzisionsbiopsien schließlich zur Diagnose einer AL-Amyloidose.
© Juan Gärtner – stock.adobe.com
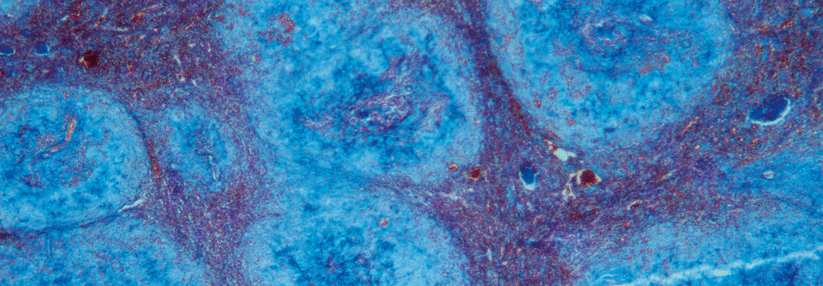
Die Bezeichnung Amyloidose fasst eine Reihe sehr heterogener Multisystemerkrankungen in einem Begriff zusammen. Allen gemeinsam ist die Bildung fehlgefalteter Proteine, sogenannter Amyloide, die sich in den Organen ablagern und deren Funktion stören, schreibt Dr. Elisabeth Blüthner von der Charité – Universitätsmedizin Berlin. In Abhängigkeit vom verursachenden Eiweiß entstehen die verschiedensten klinischen Erscheinungsbilder. Bislang sind über 40 amyloide Proteine bekannt, die sich nur anhand einer Biopsie voneinander unterscheiden lassen. Die häufigsten Formen sind die Leichtketten-, die Serumamyloid-A- und die Transthyretin-Amyloidose (AL-, AA- bzw. ATTR-Amyloidose).
Leichtketten-Amylo…
Liebe Leserin, lieber Leser, aus rechtlichen Gründen ist der Beitrag, den Sie aufrufen möchten, nur für medizinische Fachkreise zugänglich. Wenn Sie diesen Fachkreisen angehören (Ärzte, Apotheker, Medizinstudenten, medizinisches Fachpersonal, Mitarbeiter der pharmazeutischen oder medizintechnischen Industrie, Fachjournalisten), loggen Sie sich bitte ein oder registrieren sich auf unserer Seite. Der Zugang ist kostenlos.
Benutzeranmeldung
Bitte geben Sie Ihren Benutzernamen und Ihr Passwort ein, um sich an der Website anzumelden.
Bei Fragen zur Anmeldung senden Sie bitte eine Mail an online@medical-tribune.de.