Hämoglobinopathien Erwachsen werden trotz Sichelzellkrankheit
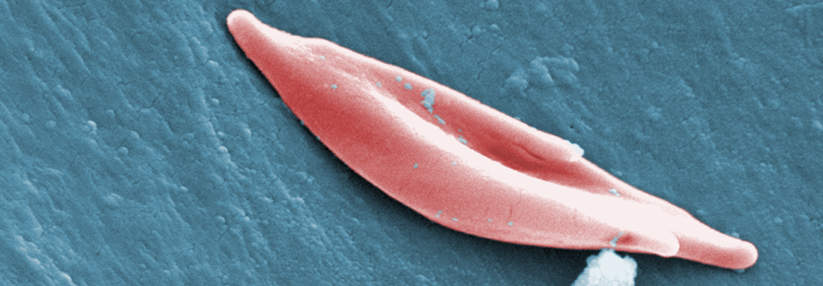
 In diesem Ausstrich sind die Sichelzellen als Folge der Polymerisierung des Hämoglobins S auf den ersten Blick zu erkennen.
© wikimedia commonas/Paulo Henrique Orlandi Mourao
In diesem Ausstrich sind die Sichelzellen als Folge der Polymerisierung des Hämoglobins S auf den ersten Blick zu erkennen.
© wikimedia commonas/Paulo Henrique Orlandi Mourao
Als Hämoglobinopathie werden Veränderungen der Struktur, Funktion oder Bildung des Blutfarbstoffs bezeichnet. Am häufigsten treten Sichelzellkrankheit (SK) und b-Thalassämie (b-Thal) auf. Beide werden autosomal rezessiv vererbt. Bei der Sichelzellkrankheit führt eine Punktmutation im b-Globin-Lokus zur Produktion eines veränderten Hämoglobins (HbS). Die SK tritt in zwei Formen auf: als homozygote HbSS und als Compound-heterozygote Störung. Bei Letzterer besteht neben der HbS-Mutation noch eine Veränderung im zweiten beta-Globin-Allel, am häufigsten ist die HbSC-SK.
Die Lebensdauer der Erythrozyten ist bei der HbSS von 120 Tagen auf etwa zehn Tage verkürzt. Exogene Faktoren wie Hypoxie, Kälte, Infektionen oder Dehydrierung lassen das HbS polymerisieren. Die Folge sind vasookklusive Krisen. Gefäßverschlüsse und Erythrozytenzerfall führen zu Entzündungen, Endothelschäden und Nekrosen, schreibt Dr. Anne Marie Asemissen vom Universitären Cancer Center Hamburg am Universitätsklinikum Hamburg-Eppendorf.
Patienten mit Sichelzellkrankheit haben typischerweise eine normozytäre Anämie mit Hämolysezeichen. Das Leitsymptom ist die Schmerzkrise. Die HbSS fällt meist bereits in der Kindheit auf, die HbSC-SK wird manchmal erst im Erwachsenenalter erkannt. Im Verdachtsfall ist eine genauere Abklärung (z.B. Hb-Elektrophorese) indiziert, vor allem bei einer Herkunft aus Regionen mit hoher Prävalenz (s. Kasten). Die genetische Diagnostik kann zwischen homozygoter und Compound-heterozygoter Form differenzieren.
Regionale Verteilung
Wegen der relativen Malariaresistenz treten evolutionsbedingt Sichelzellkrankheit und β-Thalassämie besonders häufig im Mittelmeerraum, dem subsaharischen Afrika und als Folge des Sklavenhandels auch in Amerika auf. Die Thalassämie findet sich zusätzlich in Südostasien.
Gallensteine schon im Jugendalter
Zu den führenden Symptomen der HbSC-SK zählen vasookklusivbedingte Schmerzkrisen, Hirninsult und Hörsturz. Außerdem kann es zu Milzsequestration, akutem Thoraxsyndrom und Priapismus kommen. Auch mit Endorganschäden wie pulmonaler Hypertonie, Hepatopathie, Knochenmark- und Niereninsuffizienz muss man rechnen. Weitere negative Folgen sind proliferative Retinopathie, Hüft- und Humeruskopfnekrosen sowie Osteoporose. Nicht selten bilden sich schon in der Jugend Gallensteine.
Inzwischen erreichen 90 % der Betroffenen das Erwachsenenalter, Voraussetzung: die zeitige Diagnose. Empfohlen wird eine Penicillinprophylaxe für Säuglinge und Kleinkinder sowie Impfungen gegen Pneumo- und Meningokokken. Wichtig sind jährliche Vorsorgeuntersuchungen und eine frühe Antibiotikatherapie bei Fieber. Eine weitere Verbesserung kann durch die Therapie mit Hydroxycarbamid erzielt werden. Transfusionen brauchen die Patienten unter konsequenter Behandlung nur noch selten. Eine kurative Option bietet die Stammzelltransplantation, sie sollte möglichst schon in einem Alter bis 14 Jahren mit HLA-identischen Familienspendern erfolgen.
Patienten mit Compound-heterozygoten SK leiden vor allem an Augenschäden, Hörminderung sowie Femur- und Humeruskopfnekrosen. Viele sind bereits als junge Erwachsene blind und taub. Therapeutisch profitieren sie in erster Linie von Aderlässen, die Schmerzen und andere Symptome lindern und arteriellen Thromboembolien vorbeugen.
Die b-Thalassämie als zweite wichtige Erkrankung entsteht durch eine reduzierte oder fehlende Betaglobinsynthese. Das führt zu einem Überschuss an a-Globinketten, die instabile Tetramere bilden. Infolgedessen kommt es zu intramedullärer Hämolyse, ineffizienter Erythropoese und Störungen des Eisenstoffwechsels. Statt der früher üblichen Differenzierung in eine Minor- und Majorform spricht man heute von einer transfusionsabhängigen (TDT) und nicht-transfusionsabhängigen (NTDT) Thalassämie. Bei der TDT besteht eine homozygote oder Compound-heterozygote b0-Mutation, die Träger benötigen meist ab der Geburt Blutübertragungen. Patienten mit NTDT haben b+-Mutationen. Transfusionen sind nur in kritischen Situationen erforderlich (z.B. Wachstumsphase, Schwangerschaft, Begleiterkrankungen). Die heterozygote Trägerschaft einer b°- oder b+ löst eine überwiegend asymptomatische Mikrozytose aus.
Krankheit statt Anämie
Statt von einer Sichelzellanämie spricht man heute von einer Sichelzellkrankheit. Denn die negativen Folgen werden vor allem durch Gefäßverschlüsse und Hämolysen verursacht, eher selten durch eine Anämie. Manche Formen sind gar nicht mit einer Blutarmut verbunden.
Die TDT wird bereits im Säuglings- oder Kleinkindalter erkannt. Bei Erwachsenen lässt sich die NTDT aufgrund des Blutbildes vermuten. Dabei fallen vermehrte mikrozytäre, hypochrome Erythrozyten bei allenfalls gering erniedrigtem Hb auf. Die Therapie der TDT basiert auf einem Hyperperfusionsregime, mit dem die ineffiziente Erythropoese supprimiert werden soll.
Die Symptomatik der Thalassämie selbst wird durch die Kombination von Anämie und Eisenüberladung verursacht. Die ineffektive Erythropoese führt unbehandelt zu einer extramedullären Blutbildung mit Hepatosplenomegalie, Knochendeformitäten, Fatigue und Pseudotumoren bis hin zu einer Myelonkompression mit Querschnittssymptomen. Die hämolysebedingte Endothelentzündung begünstig thromboembolische Ereignisse. Fast alle Patienten entwickeln als Jugendliche eine Cholezystolithiasis. Außerdem entstehen durch das Mischbild von ineffizienter Erythropoese und Eisenüberladung endokrine Störungen wie Diabetes und Hypothyreose.
Luspatercept reduziert den Transfusionsbedarf
Ein optimales Transfusionsregime mit Eisenchelationstherapie ab der frühen Kindheit kann diese Komplikationen verhindern. Außerdem steht mit Luspatercept inzwischen ein Medikament zur Verfügung, das die ineffektive Erythropoese verringert. In Zulassungsstudien konnte eine Reduktion des Transfusionsbedarfs gezeigt werden. Die Stammzelltransplantation ist die Therapie der Wahl bei Kindern mit TDT. Für erwachsene Patienten kommt sie in Betracht, wenn diese wegen einer Alloimmunisierung nicht langfristig transfundiert werden können.
Quelle: Asemissen AM. Hamburger Ärzteblatt 2023; 77: 12-17