
Enzymdefekt bremst Energiezufuhr – wenn Mutationen die Muskeln schwächen
 Im histologischen Bild zeigt sich beim M. Pompe eine vakuoläre Myopathie.
© wikimedia/Jensflorian (CC BY-SA 3.0)
Im histologischen Bild zeigt sich beim M. Pompe eine vakuoläre Myopathie.
© wikimedia/Jensflorian (CC BY-SA 3.0)
Glykogenosen (GSD) liegt häufig ein Defekt des lysosomalen Glykogenabbaus zugrunde, sodass Glykogen in den Lysosomen gespeichert wird. Das ist z.B. der Fall bei der GSD Typ II, auch bekannt als Morbus Pompe. Andere wiederum beruhen auf einem Defekt der anaeroben Glykogenolyse, die Speicherung des Kohlenhydrats findet dann im Zytosol statt. Ein Beispiel: die GSD Typ V (McArdle-Erkrankung), die auf einem Myophosphorylasemangel beruht.
Der Morbus Pompe manifestiert sich je nach Restaktivität des fehlenden Enzyms Alpha-1/4-Glukosidase schon im Säuglingsalter (Restaktivität < 2 %) oder später im Leben (Restaktivität 2–30 %). Bei Säuglingen tritt eine rasch progrediente, proximal betonte Muskelhypotonie („floppy infant“) mit Saug- und Trinkschwäche sowie respiratorischer Insuffizienz auf. Sehr oft liegt auch eine hypertrophe Kardiomyopathie oder eine Herzrhythmusstörung vor, schreiben Dr. Annika Saak und Dr. Jochen Schäfer von der Universitätsklinik für Neurologie, Dresden.
Jugendliche oder Erwachsene mit Morbus Pompe leiden unter einer progredienten proximalen und beinbetonten symmetrischen Gliedergürtelmyopathie mit Myalgien. Prognostisch ungünstig ist die Affektion des Zwerchfells, die zu einer respiratorischen Insuffizienz führt. Die Patienten klagen über Dyspnoe im Liegen. Viele bedürfen einer nicht-invasiven Beatmung.
Die McArdle-Erkrankung fällt in der Regel erstmals im Jugendalter durch eine Belastungsintoleranz auf. Moderate Aktivitäten tolerieren die Betroffenen besser, weil der Muskel dann die meiste benötigte Energie aus der Fettsäureoxidation bezieht.
Probleme treten bei kurzfristiger intensiver Belastung auf, wenn die Glykogenolyse angeworfen wird, kennzeichnend sind dann proximal betonte Krämpfe, Muskelschwellungen und Myalgien. Es kann auch zur belastungsinduzierten Myoglobinurie kommen (colafarbener Urin!), die das Risiko einer Rhabdomyolyse birgt. Im weiteren Verlauf der Erkrankung entwickelt sich allmählich eine proximal betonte Muskelschwäche mit Atrophie.
Vor allem eine GSD V geht mit einer CK-Erhöhung bis zum 20-Fachen der Norm einher. Bei der GSD II ist dieser Befund weniger ausgeprägt. Sichern lässt sich die Diagnose einer GSD V mit dem sogenannten Arbeitstest. Hier werden nach Kontraktion der Unterarmflexoren der Laktat- und der Ammoniak-Spiegel bestimmt. Das Laktat steigt bei einer GSD V kaum an, Ammoniak dagegen überschießend.
Pompe und McArdle molekulargenetisch sichern
Die Muskel-MRT zeigt bei der GSD II eine Signalsteigerung (Atrophie) in den Adduktoren, paravertebral, in der Glutealmuskulatur und im M. semimembranosus. Elektromyographisch findet man pathologische Spontanaktivität und ein myopathisches Muster. Die Muskelbiopsie ergibt eine vakuoläre Myopathie, Dominanz der Typ-I-Fasern und erhöhte Aktivität der sauren Phosphatase. Bei der GSD V sieht man eine subsarkolemmale Glykogenspeicherung, Atrophie von Typ-I-Fasern und eine abgeschwächte Phosphorylasereaktion. Letztlich sollte die Diagnose beider Erkrankungen durch einen molekulargenetischen Nachweis der zugrunde liegenden Mutation gesichert werden.
Allen Patienten mit Glykogenosen wird ein aerobes Ausdauertraining mit 60–70 % der maximalen Herzfrequenz empfohlen. Für die GSD II steht eine Enzymsubstitutionstherapie mit Alglukosidase alpha (20 mg/kgKG alle 14 Tage) zur Verfügung, die die respiratorische Funktion bessern kann.
Bei der McArdle-Erkrankung kann eine symptomatische Therapie mit Kreatin (60 mg/kgKG/d)erfolgen. Hilfreich ist auch, vor einer Belastung Rohrzucker oder Glukose zuzuführen. Vitamin B6, ein Kofaktor der Myophosphorylase, lohnt ebenfalls einen Versuch in einer Dosis von 60–90 mg/d.
Lipidspeichermyopathien sind klinisch und genetisch heterogene Enzymdefekte des muskulären Lipidstoffwechsels, die die zelluläre Karnitinaufnahme, die mitochondriale Fettsäureaufnahme oder -betaoxidation, den Coenzym-Q10-Stoffwechsel oder den Phosphatidsäureabbau betreffen. Es kommt vor allem zur Lipidspeicherung im Skelettmuskel. Ein primärer Karnitinmangel und Betaoxidationsdefekte präsentieren sich je nach Manifestationsalter unterschiedlich schwer.
Die Formen des primären Karnitinmangels und der Betaoxidationsdefekte
- Neonatale Form: schwerer Verlauf mit Enzephalopathie, Hypotonie, Organomegalien, Kardiomyopathie, Leberversagen, evtl. auch hypoketotische Hypoglykämie, Azidose, Epilepsie oder Dysmorphien
- Infantile Form: Reye-Syndrommit Enzephalopathie, Leberversagen, Kardiomyopathie und Hypoglykämie
- Juvenile Form: isolierte proximale Myopathie mit fixierter Schwäche oder episodisch auftretenden Myallien und Rhabdomyolyse
Eine Unterform der Lipidspeichermyopathien sind die Neutralfettspeicherkrankheiten, denen Defekte des Lipaseaktivators oder der muskulären Triglyzeridlipase zugrunde liegen. Lipide werden bei ihnen auch in Herz, Innenohr, ZNS, Haut und Leukozyten gespeichert. Die Betroffenen leiden unter einer langsam progredienten, proximal betonten, schmerzlosen Muskelschwäche.
Im Alltag zeigen viele Patienten keine Krankheitssymptome. In besonderen Situationen, zum Beispiel bei Infektionen oder nach Operationen kann es jedoch zu einer Dekompensation mit Myoglobinurie, Rhabdomyolyse und Hypoglykämie kommen. Durch die rechtzeitige Infusion von Glukoselösung lässt sich dem entgegensteuern.
Ernährung mit wenig Fett und viel Kohlenhydraten
Labordiagnostisch steht die Bestimmung von Plasma-Acylkarnitin im Mittelpunkt, schreiben die beiden Kollegen aus Dresden. Sie empfehlen, die Diagnose immer molekulargenetisch zu sichern. Die Therapie muss sich auf symptomatische Maßnahmen beschränken. Längeres Fasten oder längere körperliche Belastung gilt es zu meiden, um keine Dekompensation zu provozieren. Patienten sollten sich fettarm und kohlenhydratreich ernähren. Je nach zugrunde liegendem Enzymdefekt bietet sich die Einnahme von mittelkettigen Triglyzeriden, L-Karnitin, Riboflavin oder Coenzym Q10 an.Quelle: Saak A, Schäfer J. internistische praxis 2021; 63: 458-467
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).
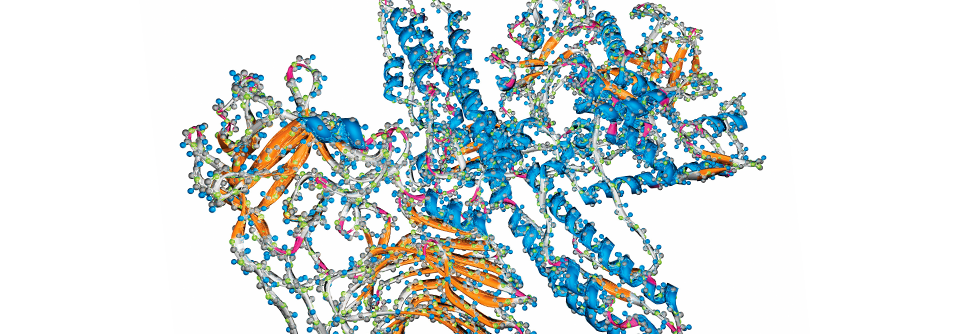



 Im histologischen Bild zeigt sich beim M. Pompe eine vakuoläre Myopathie.
© wikimedia/Jensflorian (CC BY-SA 3.0)
Im histologischen Bild zeigt sich beim M. Pompe eine vakuoläre Myopathie.
© wikimedia/Jensflorian (CC BY-SA 3.0)