
Wettlauf gegen die Zeit
 Ärzte hatten dem Jungen nur ein Jahr zu leben gegeben. Nach einer Gentherapie ist die Prognose viel besser.
© mauritius images/Tribune Content Agency LLC/Alamy
Ärzte hatten dem Jungen nur ein Jahr zu leben gegeben. Nach einer Gentherapie ist die Prognose viel besser.
© mauritius images/Tribune Content Agency LLC/Alamy
Etwa eines von 7.000 Kindern hat eine 5q-assoziierte spinale Muskelatrophie (SMA). Die Hälfte von ihnen weist bereits zum Zeitpunkt der Geburt Krankheitszeichen auf oder entwickelt diese innerhalb der ersten drei Lebensmonate. Leitsymptom ist eine generalisierte Muskelhypotonie mit proximal betonter Schwäche vorwiegend an den Beinen. Die Muskeleigenreflexe sind verringert oder fehlen. Diagnostisch wegweisende Zungenzuckungen (Faszikulationen) finden sich nur bei schweren Verläufen. Die Sensibilität bleibt erhalten und die kognitive Entwicklung verläuft normal, schreiben Dr. Burkhard Lawrenz aus Arnsberg und Professor Dr. Georg Hoffmann von der Universität Heidelberg.
Eine Genkopie sorgt für weniger aggressive Verläufe
Die autosomal-rezessiv vererbte Muskelerkrankung wird durch eine Veränderung auf dem langen Arm des Chromosoms 5 verursacht. Betroffen ist ein Gen, das für das Survival-Motoneuron-Protein kodiert (SMN1-Gen). Mehr als 95 % der Patienten weisen eine homozygote Deletion des Exon 7 auf. Anhand des Schweregrades werden fünf Formen der Erkrankung unterschieden.
| Verlaufsformen der spinalen Muskelatrophie | |||
|---|---|---|---|
| Typ I | akute infantile SMA | Symptome bereits bei der Geburt oder bis zum dritten Lebensmonat, kein freies Sitzen möglich, Tod innerhalb der ersten zwei Lebensjahre (Ateminsuffizienz, Infektionen) | 50 % der Patienten |
| Typ II | chronische infantile oder intermediäre SMA | Symptombeginn meist im 1. Lebensjahr, freies Sitzen möglich, Gehen nur mit Hilfe, Lebenserwartung eingeschränkt | 30 % der Patienten |
| Typ III | juvenile SMA | leichter Verlauf, eventuell Verlust der Gehfähigkeit im Alter, Lebenserwartung allenfalls gering eingeschränkt | 19 % der Patienten |
| Typ IV | adulte SMA | ab dem 4. Lebensjahrzehnt zunehmende Muskelschwäche bei normaler Lebenserwartung | 1 % der Patienten |
| Typ 0 | Typ 0 | Ateminsuffizienz bei der Geburt, Überlebenszeit wenige Monate | < 1 % der Patienten |
Ein vollständiges Fehlen des SMN-Proteins führt bereits innerhalb der ersten Lebensmonate zum Tod (Typ 0). Die weniger aggressiven Verläufe sind dadurch zu erklären, dass viele Patienten über Kopien eines weiteren Gens (SMN2) verfügen, das SMN1 sehr ähnlich ist. Die auf diesem Weg synthetisierte Proteinmenge ist zwar nur gering, kann aber den Untergang der Neurone verzögern und so den Verlauf abschwächen.
Für das Neugeborenen-Screening wird auf der Filterkarte eingesandtes Trockenblut genutzt. Bei homozygoten Gesunden und heterozygoten Genträgern lässt sich das Exon 7 des SMN1-Genes mittels Polymerasekettenreaktion amplifizieren. Bei Patienten mit homozygoter Deletion in diesem DNA-Abschnitt gelingt es nicht, ihr Testresultat wird deshalb als SMA-positiv gewertet.
In diesem Fall sollten Eltern und Kinder an ein neuropädiatrisches Zentrum mit Erfahrung in der SMA-Therapie überwiesen werden, wo sie innerhalb weniger Tage einen Beratungstermin erhalten. Dort erfolgen weitere Untersuchungen zur Bestätigung des Gendefekts und zur Einschätzung von Prognose und Therapieindikation.
Zu beachten ist auch, dass knapp 5 % der SMA-Patienten eine Compound-Heterozygotie mit Deletion in einem Exon 7 und Punktmutation des SMN1-Gens im anderen Allel aufweisen. Diese Konstellation wird wegen der erhaltenen Amplifikationsfähigkeit im Neugeborenen-Screening nicht erkannt. Deshalb raten die Autoren bei verdächtigen Symptomen trotz negativen Tests zur genaueren Abklärung.
Auch der Kreatinkinasewert kann in die Irre leiten. Dieser ist bei der spinalen Muskelatrophie, im Gegensatz zu den meisten anderen Muskeldystrophien, überwiegend normal oder nur geringfügig erhöht.
Psychosoziale Unterstützung
Beistand und Hilfsangebote für betroffene Familien vermitteln die Deutsche Gesellschaft für Muskelkranke, die SMA-Initiative und die Deutsche Muskelstiftung.
Mittlerweile existieren mehrere kausale Therapiemöglichkeiten für die 5q-assoziierte spinale Muskelatrophie. Eine Option ist die Steigerung der Konzentration der im SMN2-Gen codierten Proteine. Für diesen Ansatz wurde als erstes Pharmakon Nusinersen für alle Altersstufen zugelassen. Es muss regelmäßig intrathekal appliziert werden – anfangs 14-tägig, später alle vier Monate. Als Alternative kommt für Kinder ab zwei Monaten das oral einzunehmende Risdiplam in Betracht.
Auch eine Genersatztherapie steht inzwischen zur Verfügung. Der hierfür eingesetzte Wirkstoff Onasemnogen abeparvovec enthält nicht-vermehrungsfähige Adenoviren, in die rekombinante DNA des SMN1-Gens eingebracht wurde. Zur Verabreichung genügt eine einmalige einstündige Infusion. Wegen der Immunreaktion auf den Vektor brauchen die Patienten zusätzlich eine systemische Steroidbehandlung. Diese sollte in den 24 Stunden vor und mindestens 30 Tage nach der Anwendung erfolgen, zuzüglich einer Ausschleichzeit von vier Wochen.
Quelle: Lawrenz B, Hoffmann GF. Kinder- und Jugendarzt 2021; 52: 600-604
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).

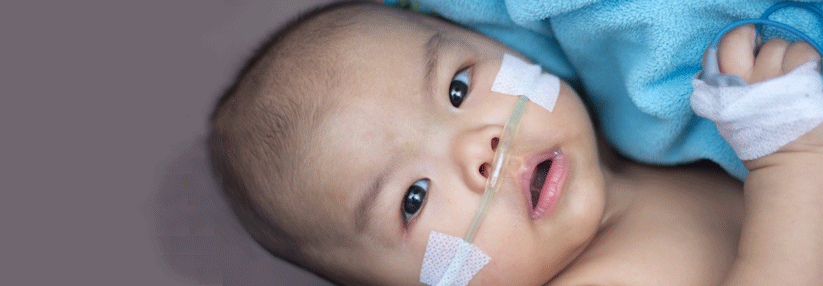



 Ärzte hatten dem Jungen nur ein Jahr zu leben gegeben. Nach einer Gentherapie ist die Prognose viel besser.
© mauritius images/Tribune Content Agency LLC/Alamy
Ärzte hatten dem Jungen nur ein Jahr zu leben gegeben. Nach einer Gentherapie ist die Prognose viel besser.
© mauritius images/Tribune Content Agency LLC/Alamy