Spinale Muskelatrophie: Behandelbar durch neue Therapieoptionen
 Bis zur Rollstuhlpflicht soll es möglichst nicht kommen. Ob man das Ziel mit den modernen Therapeutika erreichen kann, bleibt abzuwarten.
© Science Photo Library/Grala, Suzanne
Bis zur Rollstuhlpflicht soll es möglichst nicht kommen. Ob man das Ziel mit den modernen Therapeutika erreichen kann, bleibt abzuwarten.
© Science Photo Library/Grala, Suzanne
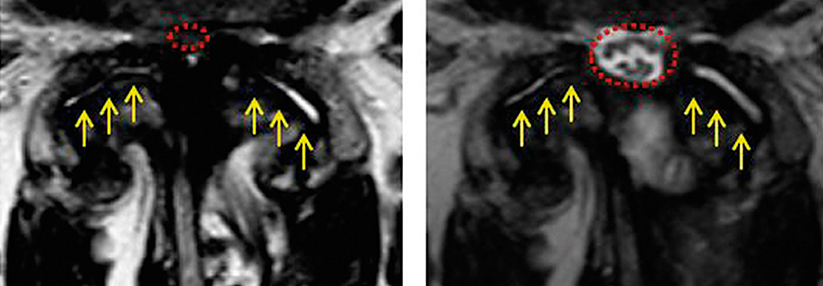
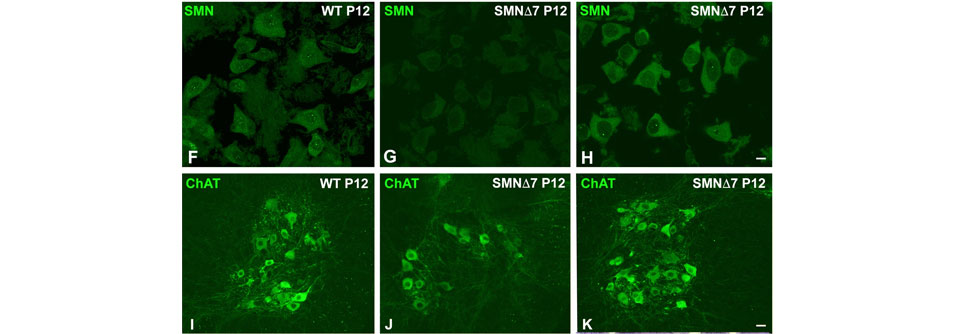
Für Patienten mit spinaler Muskelatrophie (SMA), deren Angehörige und viele Ärzte hat eine neue Zeitrechnung begonnen. Mit den beiden Therapeutika Nusinersen und Onasemnogen-Abeparvovec-Xioi kann der Erbkrankheit nun kausal begegnet werden. Aller begründeter Euphorie zum Trotz darf jedoch nicht vergessen werden, dass bisher nur wenige belastbare Daten über die Behandlungen zur Verfügung stehen – Daten, die für Diagnostik, Therapieentscheidung und Prognose unabdingbar sind, schreiben Dr. Megan A. Waldrop und Dr. Bakri H. Elsheikh vom Ohio State University Wexner Medical Center in Columbus.
Liebe Leserin, lieber Leser, aus rechtlichen Gründen ist der Beitrag, den Sie aufrufen möchten, nur für medizinische Fachkreise zugänglich. Wenn Sie diesen Fachkreisen angehören (Ärzte, Apotheker, Medizinstudenten, medizinisches Fachpersonal, Mitarbeiter der pharmazeutischen oder medizintechnischen Industrie, Fachjournalisten), loggen Sie sich bitte ein oder registrieren sich auf unserer Seite. Der Zugang ist kostenlos.
Benutzeranmeldung
Bitte geben Sie Ihren Benutzernamen und Ihr Passwort ein, um sich an der Website anzumelden.
Bei Fragen zur Anmeldung senden Sie bitte eine Mail an online@medical-tribune.de.