Jeder Vierte stirbt noch auf der Intensivstation
 Entzündlich-rheumatische Erkrankungen haben manchmal auch lebensbedrohliche kardiale
Auswirkungen.
© iStock/ Morsa Images
Entzündlich-rheumatische Erkrankungen haben manchmal auch lebensbedrohliche kardiale
Auswirkungen.
© iStock/ Morsa Images
Am häufigsten führen systemische Vaskulitiden, systemischer Lupus erythematodes (SLE) und rheumatoide Arthritis (RA) aufgrund eines Organversagens auf die Intensivstation, schreiben Dr. Florian Günther und Professor Dr. Martin Fleck von der Klinik für Rheumatologie/Klinische Immunologie, Asklepios Klinikum Bad Abbach. Diese Ergebnisse entstammen einer aktuellen retrospektiven Kohortenstudie, in die 525 Patienten mit schweren, rheumatisch bedingten Organmanifestationen eingeschlossen waren. Interessantes Detail: Bei 14,3 % von ihnen wurde die zugrunde liegende rheumatische Erkrankung erst auf der Intensivstation diagnostiziert.
Nur knapp 60 % der Patienten überleben das erste Jahr
Die Mortalität war insgesamt hoch. Sie betrug 23,8 % während der Zeit auf der Intensivstation und stieg binnen eines Jahres auf 39 %. Als Risikofaktoren für eine schlechte Prognose nennen die Autoren:
- höheres Lebensalter
- vorausgegangene Glukokortikoidtherapie
- SAPS-II* ≥ 50
- invasive Beatmung
- erforderliche Nierenersatztherapie
Schwere Organmanifestationen bei entzündlich-rheumatischen Erkrankungen betreffen vor allem Lunge, Niere, Blut und – weniger häufig – auch das Herz. An der Lunge führen sowohl pulmonale Vaskulitiden als auch interstitielle Lungenerkrankungen zu kritischen Situationen. Im Rahmen der Vaskulitiden kommt es aus kleinsten Gefäßen zu lebensbedrohlichen Blutungen im Bereich der Alveolen, was als diffuse alveoläre Hämorrhagie (DAH) bezeichnet wird. Häufigste Ursachen der autoimmun bedingten DAH sind ANCA-assoziierte Vaskulitiden, SLE und antiglomeruläre Basalmembran-Erkrankungen (anti-GBM) mit einem kombinierten Angriff auf alveoläre und glomeruläre Basalmembranen.
Klinisch zeigt die DAH eine große Bandbreite. Diese reicht von leichter Atemnot bis hin zu akuter schwerer Hypoxie, die eine invasive Beatmung oder extrakorporale Membranoxygenierung (ECMO) erforderlich macht. Bluthusten kommt vor, fehlt aber bei bis zu einem Drittel der Patienten.
Milchglasartige Veränderungen und Konsolidierungen in der CT sind Hinweise auf eine DAH. Gesichert wird die Diagnose jedoch durch die Bronchoskopie, bei der die aspirierten BAL-Fraktionen zunehmend blutiger werden. Die zytologische Untersuchung der Lavage ergibt große Mengen Hämosiderin-beladener Alveolarmakrophagen. Auch mikrobiologisch wird das Material untersucht, um entweder eine infektiöse Genese der DAH auszuschließen oder eine Superinfektion zu erkennen.
Der Verdacht auf eine entzündlich-rheumatisch bedingte DAH lässt sich durch das immunologische Labor erhärten. Je nach Grunderkrankung findet man z.B. antinukleäre Antikörper (bei SLE), Kryoglobuline (bei kryoglobulin-ämischer Vaskulitis), MPO-AK (bei mikroskopischer Polyangiitis) oder Anti-GBM-AK (bei antiglomerulärer Basalmembran-Erkrankung). Zusätzlich ist auf weitere entzündlich-rheumatische Manifestationen zu achten. Um die Diagnose abzusichern, sind manchmal je nach Klinik auch Gewebeentnahmen aus Nasenschleimhaut, Haut oder Lunge erforderlich. Bei auffälliger Urinanalyse empfiehlt sich eine Nierenbiopsie.
Therapiert wird die DAH mit hoch dosierten Glukokortikoiden (Prednisolon 250–1000 mg/d i.v.). Je nach Grunderkrankung kommen z.B. Cyclophosphamid oder Rituximab hinzu. Von einer Plasmapherese wird zunehmend abgeraten. Nur bei DAH aufgrund einer Anti-GBM-Erkrankung empfehlen die Leitlinien diese noch first line zusätzlich zu Cyclophosphamid und Steroiden.
Auch entzündlich-rheumatische interstitielle Lungenerkrankungen (ILD) können hochakut werden und innerhalb kürzester Zeit eine Beatmung erzwingen. Besonders fulminante Verläufe drohen beispielsweise bei der amyopathischen Dermatomyositis. Klinische Hinweise auf eine rheumatische Genese der ILD geben u.a. Hautveränderungen, Muskelschwäche oder Arthritiden. Diagnostisch entscheidend sind das immunologische Labor (SSc- und RA-assoziierte Antikörper, myositischspezifische oder -assoziierte AK) und die Bestimmung der Muskelenzyme. Um eine bakterielle
Superinfektion auszuschließen, empfiehlt sich zudem immer die Bronchoskopie mit BAL und mikrobiologischer Untersuchung. Therapeutisch werden meist Cyclophosphamid oder Mycophenolat- Mofetil eingesetzt, weitere Optionen sind Rituximab und Nintedanib.
Bei einer Kombination von DAH und rapid-progressiver Glomerulonephritis spricht man vom pulmorenalen Syndrom (PRS). Hier liegen meist ANCA-assoziierte Vaskulitiden oder die Anti-GBM-Erkrankung, seltener auch ein SLE zugrunde. Diagnostisch wegweisend ist neben den Zeichen der DAH das nephritische Urinsediment mit Akanthozyten und Erythrozyten-zylindern. Zur Sicherung der Diagnose dienen immunologisches Labor und Nierenbiopsie inklusive Immunhistologie. Behandelt wird das PRS wie die DAH, unter Berücksichtigung der Therapieprinzipien der zugrunde liegenden Erkrankungen.
Zwei weitere lebensbedrohliche Komplikationen rheumatisch-entzündlicher Erkrankungen sind das Makrophagenaktivierungssyndrom (MAS) und das katastrophale Antiphospholipidsyndrom (CAPS). Der für das MAS typische Zytokinsturm führt zu einem sepsisähnlichen Krankheitsbild mit therapierefraktärem Fieber, Hepatosplenomegalie und Hautveränderungen. Es droht eine Koagulopathie mit disseminierter intravasaler Gerinnung (DIC). Im Labor findet sich die typische exzessive Ferritinerhöhung neben Bi- oder Panzytopenie, erhöhten Transaminasen und erniedrigtem Fibrinogen. Unbehandelt verläuft das MAS letal. Auf kontrollierten Studien beruhende Therapieempfehlungen gibt es nicht, behandelt wird meist mit hoch dosierten Glukokortikoiden und Immunglobulinen i.v. bzw. Ciclosporin A. Neuere Therapieoptionen vor dem Hintergrund der Zytokinfreisetzung sind IL-1- bzw. IL- 6-Blocker wie Anakinra oder Tocilizumab. Bei Therapieversagen ist den Autoren zufolge ein Versuch mit Etoposid zu erwägen.
Das CAPS entwickelt sich bei etwa 1 % der Patienten mit primärem oder sekundärem
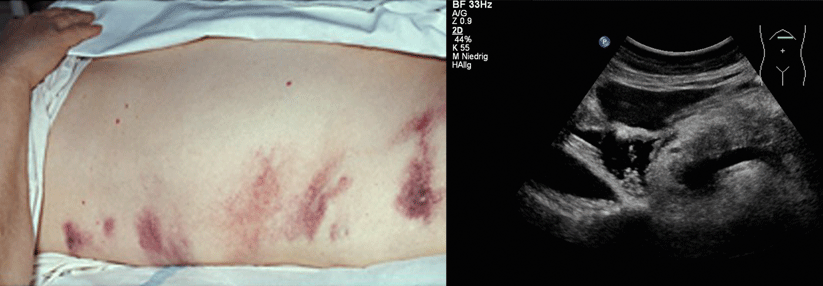
Antiphospholipidsyndrom und bedroht vor allem Niere, Lunge, Gehirn und Nebenniere. Klinisch imponieren ARDS, Enzephalopathie und akutes Nierenversagen. Labortypisch sind Antiphospholipid-AK, Thrombozytopenie, hämolytische Anämie und Zeichen der DIC. Eine schnell eingeleitete aggressive immunsuppressive und antikoagulatorische Behandlung ist essenziell, um Zytokinkaskade und Inflammation einzudämmen. Leitlinien empfehlen dafür neben Glukokortikoiden und Heparin die Plasmapherese oder alternativ intravenöse Immunglobuline. Bei therapierefraktärem Verlauf sind Rituximab und Eculizumab eine Option.
Perikardtamponade und Myokarditis durch SLE
Entzündlich-rheumatische Erkrankungen haben manchmal auch lebensbedrohliche kardiale Auswirkungen. Kollagenosen, SLE, eosinophile Granulomatose mit Polyangiitis und RA können eine Perikarditis samt Perikardtamponade auslösen. Akute Myokarditiden mit Rhythmusstörungen und Herzinsuffizienz werden vor allem beim SLE, bei eosinophiler Granulomatose mit Polyangiitis und bei systemischer Sklerose beobachtet.
Bei der Takayasu-Arteriitis (TA) sind in 10–30 % der Fälle die Koronarien beteiligt, bei Polyarteriitis nodosa autoptischen Untersuchungen zufolge sogar in bis zu 50 %. Deshalb gehen beide Erkrankungen oft mit Myokardinfarkten einher. Diese treten auch beim Antiphospholipidsyndrom auf. In letzterem Fall ist besonders zu beachten, dass nicht etwa die zur Behandlung einer Plaqueruptur übliche Stentimplantation und Plättchenhemmung, sondern die therapeutische Antikoagulation das entscheidende Therapieprinzip darstellt.
* Simplified Acute Physiology Score
Quelle: Günther F, Fleck M. Dtsch Med Wochenschr 2021; 146: 1152-1158; DOI: 10.1055/a-0949-4889
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).


 Entzündlich-rheumatische Erkrankungen haben manchmal auch lebensbedrohliche kardiale
Auswirkungen.
© iStock/ Morsa Images
Entzündlich-rheumatische Erkrankungen haben manchmal auch lebensbedrohliche kardiale
Auswirkungen.
© iStock/ Morsa Images