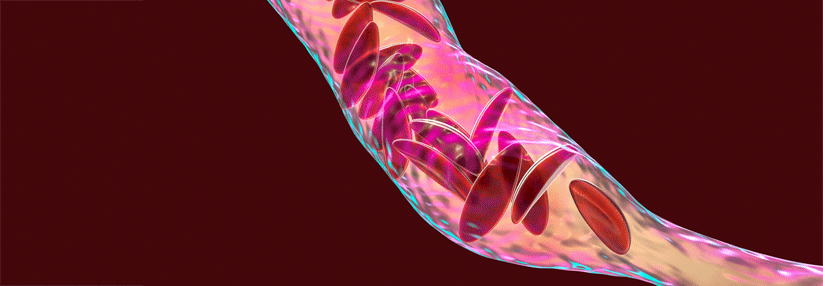
Sichelzellkrankheit wird in Deutschland häufiger
 Gesunde, runde Erys und Sichelzellen unter dem Rasterelektronenmikroskop.
© Science Photo Library/Murti, Dr. Gopal
Gesunde, runde Erys und Sichelzellen unter dem Rasterelektronenmikroskop.
© Science Photo Library/Murti, Dr. Gopal
Bei der Sichelzellkrankheit wird aufgrund eines Aminosäureaustauschs im β-Globin-Locus auf Chromosom 11 Sichelzellhämoglobin (HbS) synthetisiert. Häufige Genotypen sind die homozygote HbSS-Sichelzellkrankheit, die HbS-β-Thalassämie und die HbSC-Krankheit, schreiben Dr. Laura Distelmaier von der Klinik für Innere Medizin am Vivantes Klinikum Neukölln in Berlin und Kollegen. Es gibt weitere, seltenere Kombinationen, bei denen auf einem Allel die HbS-Anomalie liegt und auf dem anderen Allel eine andere Variante des β-Globin-Locus. Der Begriff Sichelzellkrankheit umfasst demnach eine ganze Gruppe von Erkrankungen.
Klinisch symptomatisch werden betroffene Patienten erst nach dem zweiten Lebensmonat, da dann das fetale Hämoglobin (HbF) abfällt. Die Erkrankung wird in Deutschland oft erst spät diagnostiziert, u.a. weil es kein Neugeborenen-Screening von Risikogruppen gibt. Eine frühzeitige Diagnostik und interdisziplinäre Behandlung können jedoch die Prognose betroffener Patienten positiv beeinflussen.
Wie entstehen die Symptome der Sichelzellkrankheit?
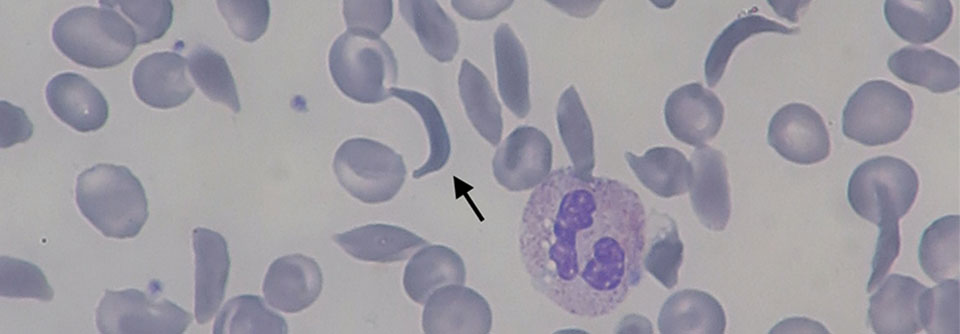
- HbS präzipitiert in desoxygeniertem Zustand, die Erythrozyten nehmen Sichelform an und verlieren ihre Verformbarkeit.
- Dies zieht eine Hämolyse nach sich und es kann zum Verschluss kleiner Gefäße kommen.
- Akut treten Gewebeischämien mit Organ- oder Knocheninfarkten auf.
- Langfristig werden Organschäden und Gefäßveränderungen beobachtet.
Im Blutbild von Patienten mit Sichelzellkrankheit zeigt sich meist eine normozytäre hämolytische Anämie mit einem Hb-Wert von 6–10 g/dl. Bei manchen Varianten der Sichelzellkrankheit wird ein Hb-Wert von 12 g/dl und mehr beobachtet. Die Leukozyten- und Thrombozytenzahlen sind oft erhöht. Zur sicheren Diagnostik sind eine Hb-Analyse und ein Hb-Löslichkeitstest erforderlich. Unter Umständen müssen auch molekulardiagnostische Untersuchungen erfolgen, beispielsweise nach vorangegangener Transfusion.
Als Auslöser für akute Komplikationen der Sichelzellkrankheit, die daher vermieden werden sollten, gelten:
- Unterkühlung
- Infekte
- Exsikkose
Jeder Vierte erleidet bis zum 45. Lebensjahr einen Apoplex
Das akute Thoraxsyndrom zählt zu den häufigsten Todesursachen von erwachsenen Patienten mit Sichelzellkrankheit. Durch Auslöser wie Infekte, Fett- oder Thromboembolien kommt es zu einer Sequestration von Blut in den Lungengefäßen. Behandelt wird überwiegend supportiv mit Sauerstoffgabe, Antibiotika und ggf. Transfusionen oder Austauschtransfusionen.Transfusionen ja oder nein?
Bluttransfusionen können bei Sichelzellkrankheit in bestimmten Situationen lebensrettend sein, aber sie bergen auch Risiken wie Alloimmunisierung, Hyperviskosität und langfristig eine Eisenüberladung.
Daher sollte man die Indikation zur Transfusion zurückhaltend stellen. Der Hb-Wert darf nicht über 10 g/dl, Hämatokrit nicht über 30 % steigen, Hyperviskosität ist zu vermeiden. Zudem wird eine erweiterte Blutgruppentestung empfohlen (Rhesusfaktor, Kell-System).
Zerebrale Ischämien und sekundäre Hämorrhagien kommen bei Patienten mit Sichelzellkrankheit gehäuft vor: Bis zum 45. Lebensjahr erleiden 24 % einen Apoplex. Zur Sekundärprophylaxe verabreicht man dauerhaft partielle Austauschtransfusionen mit dem Ziel, den HbS-Anteil im Blut unter 30 % zu halten. Eine Therapie mit Hydroxycarbamid senkt das Schlaganfall- und das Rezidivrisiko.
Weniger Komplikationen unter Hydroxycarbamid
Mit fortschreitendem Alter kommt es zunehmend zu chronischen Komplikationen wie restriktiver Lungenerkrankung, pulmonaler Hypertonie, restriktiver Kardiomyopathie, Ischämien, Infarkten, Niereninsuffizienz, Gallensteinen, Hörverlust u.v.m., die eine interdisziplinäre Therapie erfordern. Welche Behandlungsmöglichkeiten gibt es außer der Transfusion (s. Kasten)? Hydroxycarbamid senkt das Risiko akuter und chronischer Komplikationen bei mehr als zwei Drittel der Patienten. Es erhöht die Produktion von fetalem Hämoglobin und entfaltet weitere positive Effekte wie Nephroprotektion und Herunterregulation der Hyperkoagulation. Neue Medikamente und Therapieansätze wie die gentherapeutische Korrektur der HbS-Anomalie sind in Erprobung.Quelle: Distelmaier L et al. Internist 2020; 61: 754-758; DOI: 10.1007/s00108-020-00822-z
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).
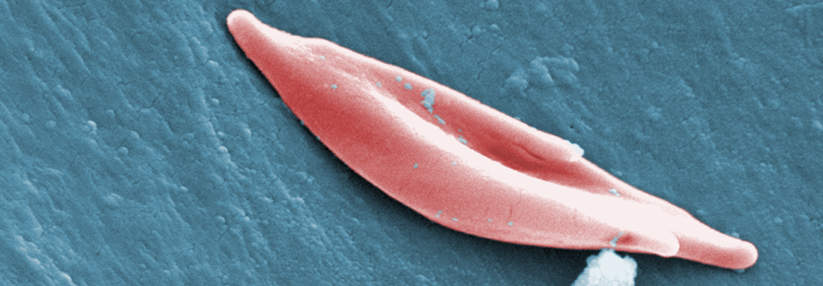
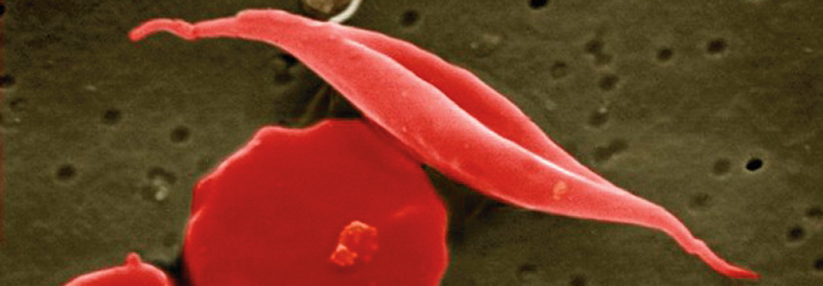



 Gesunde, runde Erys und Sichelzellen unter dem Rasterelektronenmikroskop.
© Science Photo Library/Murti, Dr. Gopal
Gesunde, runde Erys und Sichelzellen unter dem Rasterelektronenmikroskop.
© Science Photo Library/Murti, Dr. Gopal