Mukoviszidose: Von Kombinations- und Gentherapien bis hin zu neuen Antiinfektiva
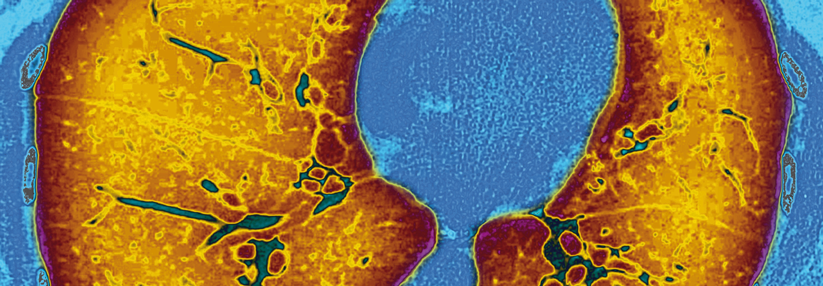
 In der Behandlung von Patienten mit zystischer Fibrose ließen sich durch CFTR-Potenziatoren sowie CFTR-Korrektoren bereits große Fortschritte erzielen.
© Science Photo Library/St. Bartholomew‘s Hospital
In der Behandlung von Patienten mit zystischer Fibrose ließen sich durch CFTR-Potenziatoren sowie CFTR-Korrektoren bereits große Fortschritte erzielen.
© Science Photo Library/St. Bartholomew‘s Hospital
Die Einführung der CFTR-Modulatoren hat die Therapie der zystischen Fibrose (CF) enorm bereichert. Potenziatoren wie Tezacaftor/Ivacaftor verlängern die Kanalöffnungszeit, Korrektoren wie Lumacaftor oder Elexacaftor sorgen für eine bessere Ausstattung der Zelloberfläche mit Kanälen. Kombiniert entfalten beide Ansätze ein beträchtliches therapeutisches Potenzial in Sachen Lungenfunktion und Exazerbationsrisiko, erklärte Dr. Damian Downey, Queen’s University Belfast.
Vor allem Staph. aureus nicht unterschätzen
Neue Studien haben gezeigt, dass eine Tripletherapie mit einem Korrektor und zwei Potenziatoren (Elexacaftor/Tezacaftor/Ivacaftor) für einen weiteren deutlichen Zugewinn an FEV1 sorgt. Zudem werden diverse Ansätze auf genetischer Ebene geprüft, um die Produktion des unreifen Kanalproteins zu steigern.
Auf die CFTR-Funktion zielende Therapien sind nicht das Ende der Fahnenstange, erinnerte Dr. Downey. „Wir sehen eine starke Pipeline von antiinfektiösen, antiinflammatorischen und die mukoziliäre Clearance verbessernden Therapien.“
Die antiinfektiöse Behandlung hat bislang vor allem Pseudomonas im Blick, aber man sollte andere Pathogene nicht unterschätzen, insbesondere Staphylococcus aureus, erklärte Professor Dr. Patrick Flume, Medical University of South Carolina in Charleston. Im amerikanischen CF-Register fällt eine hohe Nachweisrate von Staph. aureus und MRSA ins Auge, vor allem bei jüngeren Patienten bis 25 Jahren wird Staph. aureus häufiger nachgewiesen als Pseudomonas. „Die Frage ist legitim, ob wir nicht auch diese anderen Pathogene bei unseren Eradikations- und Suppressionstherapien zum Ziel machen sollten“, so Prof. Flume.
Bisher gibt es zwar weder in den USA noch in Europa ein zugelassenes inhalatives Antibiotikum für diese Indikation, aber einiges ist in der Forschung. So wird derzeit Vancomycin-Puder in einer Phase-3-Studie getestet und geprüft, ob inhalatives Fosfomycin/Tobramycin auch gegen S. aureus wirkt. Auch die laufende placebokontrollierte Phase-2-Studie mit NO, deren primärer Endpunkt die Veränderung der FEV1 ist, untersucht, wie sich die bakterielle Besiedlung der Lunge u.a. mit Pseudomonas und S. aureus entwickelt.
Akute Exazerbationen stellen bei der zystischen Fibrose ein besonderes Problem dar, weil sie viele Patienten unwiederbringlich Lungenfunktion kosten. Nur etwa die Hälfte der Betroffenen erholt sich vollständig, die Gründe dafür sind unklar. Diskutiert wird, ob eine optimierte Antibiose die Erfolgsraten der Therapie verbessern kann. Auf dieser Grundlage startete die STOP2-Studie, in der die Response von knapp 1000 Patienten mit CF-Exazerbation sieben bis zehn Tage nach Beginn der intravenösen antibiotischen Therapie geprüft wurde. Als frühe robuste Response galt, wenn die FEV1 um mindestens 8 % zugenommen hatte und der Symptomscore CRISS um mindestens elf Punkte gesunken war.
Patienten mit früher robuster Response (ERR) wurden nach der Randomisierung entweder 10 oder 14 Tage weiterbehandelt, diejenigen ohne ERR 14 oder 21 Tage. Die Studie ist abgeschlossen, die Ergebnisse stehen aber noch aus. Die Hypothese lautet, dass man Patienten mit ERR vier Tage Therapie inklusive potenzieller Nebenwirkungen ersparen kann, Patienten ohne ERR aber davon profitieren, wenn sie länger Antibiotika erhalten.
In-vitro-Resistenztest nicht ausreichend zuverlässig
Eine internationale Arbeitsgruppe, der auch Prof. Flume angehört, hat sich des Themas Antibiotikaresistenz angenommen. Schließlich hat es bei Patienten, die so häufig Antibiotika erhalten wie diejenigen mit CF, besondere Relevanz. Derzeit gibt es wenig Evidenz, dass sich die Response eines CF-Patienten anhand von In-vitro-Testungen vorhersagen lässt. Das gilt sowohl für die Keimeradikation als auch für die Exazerbationstherapie. Alternative Testmethoden haben bisher nicht überzeugen können, sodass dringend nach neuen Wegen gesucht wird. Deshalb lautet die Empfehlung der Arbeitsgruppe zwar, jährlich und im Falle einer akuten Exazerbation die Resistenz zu testen. Als Leitschnur für die Therapie sollten aber Bakterienspezies und klinische Response dienen. Die Resistenztestergebnisse werden gebraucht, wenn die klinische Response unbefriedigend bleibt.
Quelle: ERS* International Congress virtual (*European Respiratory Society)
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).






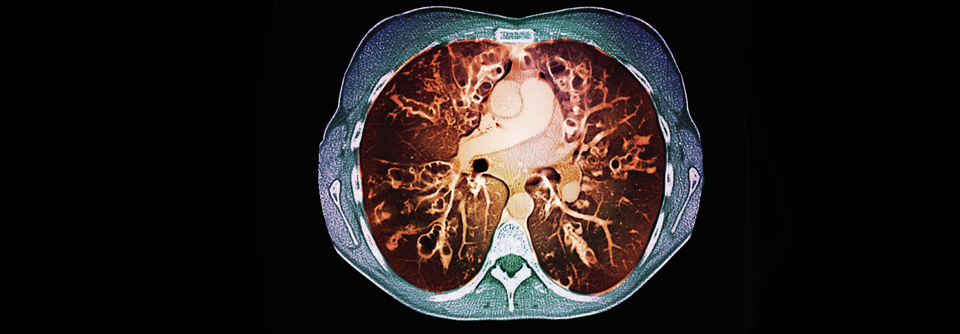


 In der Behandlung von Patienten mit zystischer Fibrose ließen sich durch CFTR-Potenziatoren sowie CFTR-Korrektoren bereits große Fortschritte erzielen.
© Science Photo Library/St. Bartholomew‘s Hospital
In der Behandlung von Patienten mit zystischer Fibrose ließen sich durch CFTR-Potenziatoren sowie CFTR-Korrektoren bereits große Fortschritte erzielen.
© Science Photo Library/St. Bartholomew‘s Hospital