Mukoviszidose Nach den CFTR-Modulatoren ist es Zeit für präzise Eingriffe am Genom
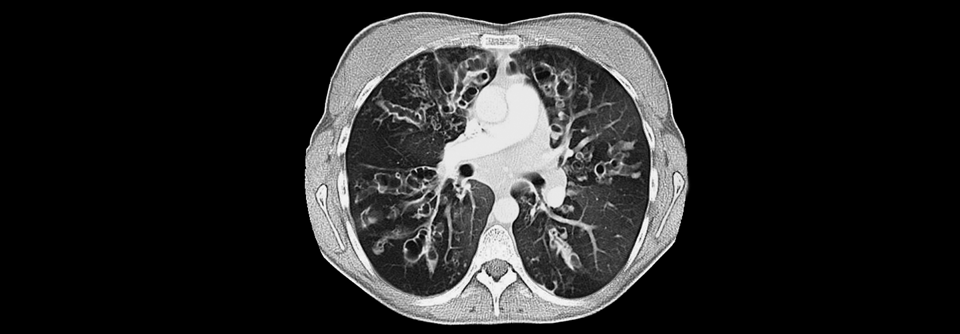
 Auf dem Thorax-Scan sieht man deutlich die abnorme Schleimproduktion (grau).
© Science Photo Library/Zephyr
Auf dem Thorax-Scan sieht man deutlich die abnorme Schleimproduktion (grau).
© Science Photo Library/Zephyr
Die Entdeckung des CFTR*-Gens im Jahr 1989 hat den Grundstein für ein besseres Verständnis der zystischen Fibrose (CF) gelegt. Demnach ist eine CFTR-Dysfunktion verantwortlich für die Kaskade aus vermehrtem Schleim in den Atemwegen, chronischen bakteriellen Infektionen und Inflammation, die diese progressive erblich bedingte Erkrankung ausmacht. Die neuen Erkenntnisse um die Pathogenese führten zunächst zu Therapien, die in diese Symptomkaskade eingreifen und z.B. die Mukus-Clearance verbessern oder die Bakterienlast reduzieren.
Hocheffektive CFTR-Modulatoren, die gezielt an den zugrundeliegenden Veränderungen ansetzen, gibt es erst seit wenigen Jahren, schreiben Dr. Simon Gräber und…
Liebe Leserin, lieber Leser, aus rechtlichen Gründen ist der Beitrag, den Sie aufrufen möchten, nur für medizinische Fachkreise zugänglich. Wenn Sie diesen Fachkreisen angehören (Ärzte, Apotheker, Medizinstudenten, medizinisches Fachpersonal, Mitarbeiter der pharmazeutischen oder medizintechnischen Industrie, Fachjournalisten), loggen Sie sich bitte ein oder registrieren sich auf unserer Seite. Der Zugang ist kostenlos.
Benutzeranmeldung
Bitte geben Sie Ihren Benutzernamen und Ihr Passwort ein, um sich an der Website anzumelden.
Bei Fragen zur Anmeldung senden Sie bitte eine Mail an online@medical-tribune.de.