
Purpura, Arthralgien und Nephritis
 Hintergrund der Vaskulitis ist die veränderte Glykosylierung von Immunglobulin A1 zu einem galaktosedefizienten IgA1 (Gd-IgA1).
© MQ-Illustrations - stock.adobe.com
Hintergrund der Vaskulitis ist die veränderte Glykosylierung von Immunglobulin A1 zu einem galaktosedefizienten IgA1 (Gd-IgA1).
© MQ-Illustrations - stock.adobe.com
Im Kindesalter verläuft die unter dem Namen Purpura Schönlein-Henoch bekannte Immunglobulin-A-Vaskulitis (IgAV) meist blande und selbstlimitierend. Nicht so bei den Erwachsenen. Bei ihnen sind häufig mehrere Organsysteme betroffen, zudem rezidiviert die Erkrankung oft und neigt dazu, chronisch zu werden, schreibt Prof. Dr. Sabine Adler vom Kantonsspital Aarau. Männer sind von der IgAV eher betroffen als Frauen, das Haupterkrankungsalter liegt zwischen dem 40. und dem 50. Lebensjahr.
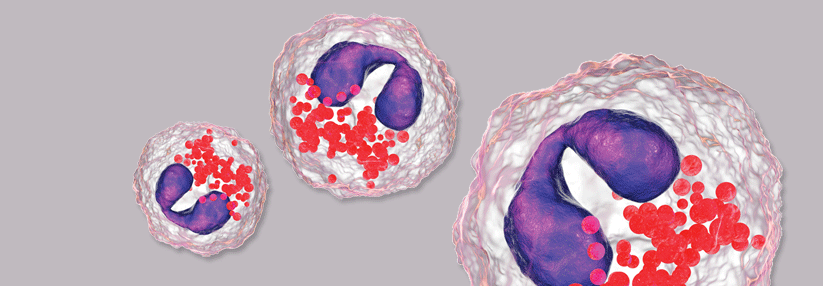
Hintergrund der Vaskulitis ist die veränderte Glykosylierung von Immunglobulin A1 zu einem galaktosedefizienten IgA1 (Gd-IgA1). Dadurch werden an dem Protein Bindungsstellen für Autoantikörper freigelegt, was wiederum eine Immunkomplexreaktion ermöglicht. Vermutlich ist die Gd-IgA1-Bildung Folge einer veränderten mukosalen Immunität, beispielsweise nach Infekten der oberen Luftwege oder des Magen-Darm-Trakts. Schlussendlich führt die Immunkomplexbildung zu perivaskulären Ablagerungen und einer Aktivierung neutrophiler Granulozyten. Diese Vorgänge können verschiedene Organsysteme betreffen und ein buntes Bild an Manifestationen ergeben.
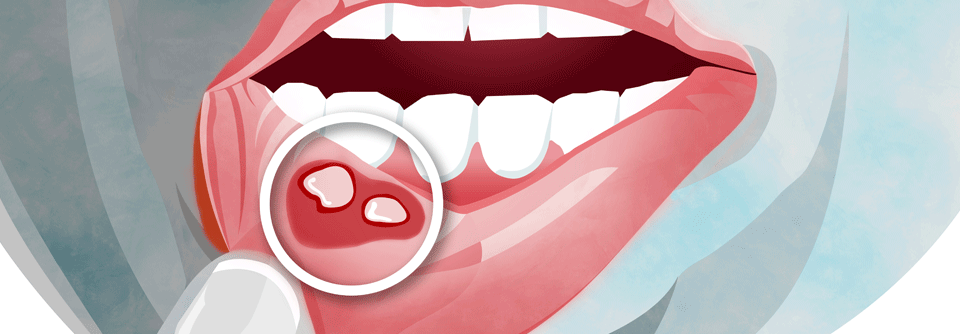
Hauptsymptom der IgAV sind palpable Purpura mit subkutaner Ödembildung – i.d.R. an den Unterschenkeln. Sie finden sich bei 70 % der Patienten schon im Initialstadium und später bei nahezu allen Betroffenen. Purpura oberhalb der Hüfte sprechen für einen generalisierten Befall, der ein erhöhtes Risiko für einen schweren Verlauf inklusive gastrointestinaler und renaler Beteiligung bedeutet.
Viele Patienten leiden unter abdominellen Krämpfen
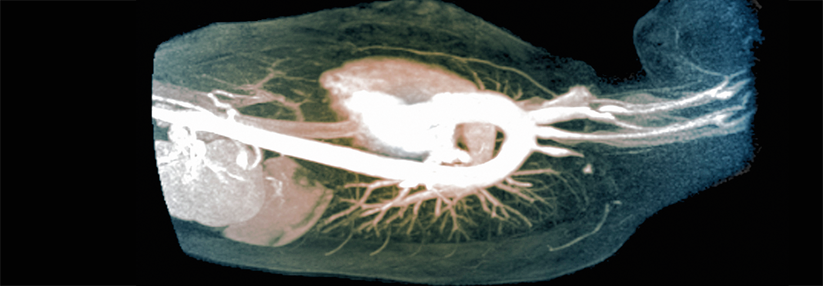
Zwei Drittel der Betroffenen leiden an Arthralgien, insbesondere der Knie- und Sprunggelenke. Schwellungen sind häufig, trotz möglicher Synovitiden kommt es bei der IgAV jedoch nicht zu destruierenden Gelenkentzündungen. Bei 50–80 % der Patienten treten gastrointestinale Beschwerden auf, vor allem in Form von krampfartigen Schmerzen. Auch Hämorrhagien sind möglich. Sie können von milden Formen mit etwas Blut im Stuhl bis zu lebensbedrohlichen Zuständen reichen. Eine zügige Diagnostik ist unumgänglich, auch um Perforationen oder Obstruktionen auszuschließen. In Sonografie und CT lassen sich oft intestinale Wandverdickungen erkennen, endoskopisch sind mukosale Erytheme und petechiale Blutungen sichtbar.
IgAV-Kriterien von EULAR, PRINTO und PreS*
Die Kriterien für die Diagnose der IgAV wurden für Kinder aufgestellt. Bei Erwachsenen mit IgAV wurden sie bisher zwar nie validiert, haben sich aber in der Praxis bewährt. Als wahrscheinlich gilt die IgAV beim Vorliegen einer typischen Purpura plus mindestens einem weiteren Kriterium:
- abdominelle Schmerzen
- IgA-Ablagerungen in der histologischen Untersuchung von Haut/Niere
- Arthritis/Gelenkschmerz
- eingeschränkte Nierenfunktion
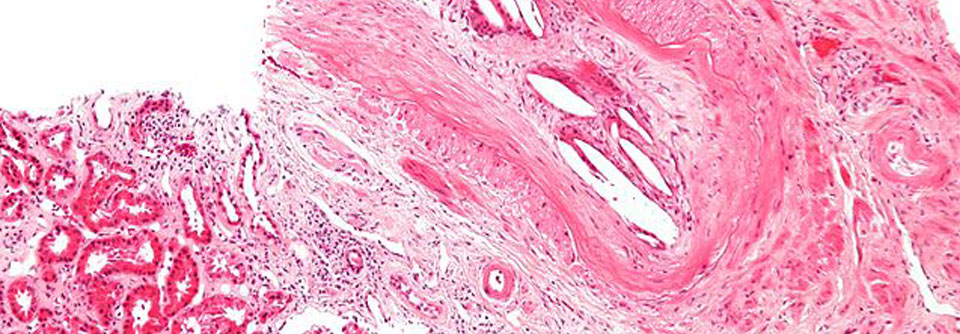
Bei etwa 70 % der erwachsenen IgAV-Patienten kommt es zu einer Nierenbeteiligung. Von asymptomatischer Mikrohämaturie bis zur terminalen Insuffizienz sind alle Ausprägungen möglich. Neben der Bestimmung von Serumkreatinin und der (Mikro-)Albuminuriediagnostik ist das Urinsediment relevant. Glomeruläre Erythrozyten weisen auf eine renale Vaskulitis hin. Wann eine Biopsie erforderlich ist, wird unterschiedlich beurteilt. Laut Prof. Adler ist die Indikation spätestens bei einer Verschlechterung der Nierenfunktion gegeben. Histologisch dominiert meist eine Immunkomplexablagerung ähnlich wie bei anderen IgA-Nephropathien.
Herzbeteiligungen sind bei der IgAV möglich, aber selten. Beschrieben wurden u.a. Palpitationen, Tachykardie, Perikardergüsse und Myokarditiden. Vermutet wird eine koronare Vaskulitis als pathophysiologischer Hintergrund. Treten im Rahmen einer IgAV kardiopulmonale Symptome auf, sollte man diesen daher unbedingt nachgehen. Bei IgAV-Patienten ohne entsprechende Beschwerden gehört die kardiale Diagnostik dagegen nicht zur erforderlichen Routine.
Die Diagnose der IgAV erfolgt anhand von Anamnese und klinischer Konstellation. Hellhörig werden muss man bei Erwachsenen, die eine Purpura ungeklärter Ursache aufweisen. In diesen Fällen kann eine Hautbiopsie helfen, die Erkrankung von anderen Vaskulitiden abzugrenzen. In der direkten Immunfluoreszenzuntersuchung lassen sich die IgA-Ablagerungen gut nachweisen, histologisch sieht man meist eine leukozytoklastische Vaskulitis.
Was fehlt, sind verlässliche serologische Marker, schreibt Prof. Adler. Bei der Hälfte der Betroffenen ist auch das namensgebende IgA im Blut unauffällig und diagnostisch nicht verwertbar. Eine mögliche Komplementerniedrigung ist zwar nicht spezifisch, nützt aber bei der Abgrenzung zu anderen Kleingefäßvaskulitiden.
Klassifikationskriterien hat man für Erwachsene ebenfalls nicht, deshalb werden diejenigen für Kinder herangezogen (siehe Kasten). Das Kriterium „eingeschränkte Nierenfunktion“ erschwert Prof. Adler zufolge jedoch die frühe Diagnose, weshalb sie es kritisch betrachtet. Besser wäre ihrer Meinung nach stattdessen das Kriterium „Urin: glomeruläre Erythrozyten plus/minus auffällige Albuminurie“.
Symptomatisch behandeln reicht oft bei mildem Verlauf
Empfehlungen zur Therapie beruhen auf Expertenmeinungen, klare Daten zur optimalen Behandlung der IgAV gibt es bisher nicht. Bei milden Verläufen kann es ausreichen, Purpura und Arthralgien symptomatisch anzugehen. Gegen die Schmerzen sollten eher Metamizol oder Paracetamol als NSAR eingesetzt werden.
Ansonsten wird stadiengerecht therapiert. Bei persistierender Proteinurie empfehlen sich RAAS-Blocker und SGLT2-Hemmer. Glukokortikoide (oral oder i.v.), Azathioprin oder MMF kommen je nach Ausmaß der Nierenbeteiligung zum Einsatz. Cyclophosphamid ist bei rasch progredienter Glomerulonephritis eine Option. Bei 22 Erwachsenen mit refraktärem Verlauf hatte auch Rituximab einen guten Effekt auf die renale Verschlechterung.
Insgesamt verläuft die IgAV oft undulierend und rezidivierend, betont Prof. Adler. In einem Kollektiv von 260 Patienten mit überwiegend mäßiger Ausprägung kam es immerhin bei 15 % der Betroffenen zu einem Rezidiv, vor allem, wenn sie keine Glukokortikoide als Basismedikation erhalten hatten. Das Gesamtüberleben betrug in dieser Kohorte 78 % nach fünf und 65 % nach zehn Jahren.
Eine Nierenbeteiligung kann im Verlauf der IgAV durchaus zeitverzögert auftreten. Um sie frühzeitig zu erkennen, brauchen IgAV-Patienten eine dauerhafte medizinische Betreuung – auch wenn sie (zunächst) nur unter milden kutanen oder rheumatologischen Manifestationen leiden. Denn die Prognose der IgAV wird vor allem von der Niere bestimmt. Bisher war die Entwicklung zur terminalen Organinsuffizienz häufig – allerdings hoffen die Experten, dass sich nach der Einführung der kombinierten RAAS/SGLT2-Hemmung das renale Outcome auch bei der IgAV verbessert hat.
Quelle: Adler S. Innere Medizin 2024; 65: 114-121, DOI:10.1007/s00108-023-01650-7
* EULAR: European Alliance of Associations for Rheumatology, PRINTO: Paedeatric Rheumatology European Society, PreS: Paediatric Rheumatology International Trials Organisation
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).


 Hintergrund der Vaskulitis ist die veränderte Glykosylierung von Immunglobulin A1 zu einem galaktosedefizienten IgA1 (Gd-IgA1).
© MQ-Illustrations - stock.adobe.com
Hintergrund der Vaskulitis ist die veränderte Glykosylierung von Immunglobulin A1 zu einem galaktosedefizienten IgA1 (Gd-IgA1).
© MQ-Illustrations - stock.adobe.com