Non-IPF-Lungenfibrose: Die Progredienz mit Antifibrotika aufhalten?
 Nicht nur bei der IPF kommt es zur progredienten Lungenfibrose.
© Science Photo Library/Alvin Telser
Nicht nur bei der IPF kommt es zur progredienten Lungenfibrose.
© Science Photo Library/Alvin Telser
Der Protoyp der progressiven fibrosierenden interstitiellen Lungenerkrankung ist die idiopathische Lungenfibrose (IPF). Doch es gibt noch weitere interstitielle Lungenerkrankungen (ILD), die jeweils in einer Subgruppe von Patienten mit zunehmender Fibrosierung einhergehen. Dazu gehören u.a. die chronische Hypersensitivitäts-Pneumonitis, mit Autoimmunerkrankungen assoziierte ILD sowie die idiopathische unspezifische interstitielle Pneumonie (NSIP). In den meisten, aber nicht allen Fällen lassen sie sich durch eine immunsuppressive Therapie stabilisieren oder sogar bessern.
Antifibrotische Medikamente wie Pirfenidon und Nintedanib mindern den jährlichen Verlust an forcierter Vitalkapazität (FVC) bei Patienten mit IPF um etwa 50 %. Die Hypothese, dass dies auch bei anderen Formen der fibrosierenden ILD funktioniert, wurde im vergangenen Jahr für Nintedanib bei Sklerodermie-assoziierter ILD bestätigt. Weitere Studien brachten Evidenz für eine Wirksamkeit dieser Substanz gegen mehrere ILD-Varianten, die trotz Erhaltungstherapie progredient verliefen.
In der Studie INBUILD, an der Patienten mit jeglicher Form einer progressiven ILD abseits der IPF teilnahmen, sank die FVC unter Nintedanib in 52 Wochen um 81 ml ab, unter Placebo um 188 ml – unabhängig von der zugrunde liegenden Pathologie. Weniger konsistent ist die Datenlage für Pirfenidon.
Auf dem dritten International Summit for Interstitial Lung Diseases haben ILD-Experten einen Konsensus zur vorliegenden Evidenz erarbeitet. Um die Basis für ein erfolgreiches Management von ILD zu schaffen, fassten Dr. Peter M. George von der Interstitial Lung Disease Unit am Royal Brompton and Harefield NHS Foundation Trust in London und Mitarbeiter die wichtigsten Ergebnisse nun in einem Positionspapier zusammen.
6-Minuten-Gehtest und DLCO ebenfalls nutzen
Wegweisend für die Definition einer progredienten Fibrose waren die Einschlusskriterien der INBUILD-Studie:
- FVC-Abfall ≥ 10 % des Sollwerts in den vorangegangenen 24 Monaten
- relativer FVC-Abfall zwischen 5 % und 10 % und Zunahme der respiratorischen Beschwerden in den vorangegangenen 24 Monaten
- relativer FVC-Abfall zwischen 5 % und 10 % sowie Zunahme der Fibrosierung in der HRCT
- Zunahme klinischer Symptome und Zunahme der Fibrosierung in der HRCT
Zusätzliche Informationen liefern der Abfall der Diffusionskapazität für Kohlenmonoxid (DLCO, vor allem in Verbindung mit einem FVC-Verlust) und die 6-Minuten-Gehstrecke, deren Rückgang allerdings oft auch andere Gründe hat. Um die Diagnose im klinischen Alltag stellen zu können, geben die Autoren klare Merkmale an die Hand (s. Kasten unten).
Kriterien der progressiven Fibrose
Die Diagnose kann nach 24 Monaten gestellt werden, wenn es trotz Therapie zu folgenden Befunden gekommen ist:
- relativer Abfall der FVC um mindestens 10 %
- relativer Abfall der FVC um mindestens 5 % plus Abfall der DLCO um mindestens 15 %
- relativer Abfall der FVC um mindestens 5 % plus zunehmende Fibrose im HRCT
- relativer Abfall der FVC um mindestens 5 % plus Progression der Symptome
- Progression der Symptome plus zunehmende Fibrose in der hochauflösenden CT
In Fällen fortschreitender Fibrose kann eine Kombination aus Immunsuppressiva und Nintedanib oder Pirfenidon bei allen ILD-Formen außer der IPF als tolerabel und sicher gelten. Eine andere Alternative wäre die Eskalation von oralen zu intravenösen Immunsuppressiva. Wie intensiv die Immunsuppression gestaltet werden kann, hängt auch ab von Alter, Komorbiditäten und Nebenwirkungen. Patienten mit IPF droht unter immunsuppressiver Therapie ein schlechteres Überleben, wozu bei ihnen auch die Telomer-Dysfunktion beiträgt.
Quelle: George PM et al. Lancet Respir Med 2020; 8: 925-934; DOI: 10.1016/S2213-2600(20)30355-6
Es wäre wünschenswert, eine Fibrose schon zu einem frühen Zeitpunkt als progredient zu definieren, um rasch Medikamente dagegen einsetzen zu können. Denn gelingt es z.B. bei der chronischen Hypersensitivitäts-Pneumonitis oder der NSIP nicht, die Krankheit in den ersten 6–12 Monaten zu stabilisieren, lässt sich der fortschreitende Lungenfunktionsverlust kaum mehr aufhalten. Die Mortalität steigt.
Wenn nach genauer Diagnose der ILD-Form eine adäquate Therapie eingeleitet wurde, raten die Auotren dazu, den Effekt auf die Lungenfunktion bereits nach drei Monaten zu überprüfen. Wie häufig eine Fibrose trotz Therapie progredient verläuft, kann man nur retrospektiv beurteilen. Aus entsprechenden Daten geht hervor, dass dies bis zu ein Drittel der Patienten betrifft. Einige Befunde gelten als Marker für eine schlechte Prognose (s. Kasten oben). Dazu gehört die Telomer-Dysfunktion, die die Prognose nicht nur bei der IPF erheblich verschlechtert. Nach Ansicht der Konsensus-Autoren sollten Telomer-Untersuchungen in der Routine häufiger erfolgen, da Patienten mit einer Dysfunktion einer engmaschigeren Überwachung bedürfen. Außerdem haben Menschen mit verkürzten Telomeren ein höheres Risiko für schwere Nebenwirkungen von Immunsuppressiva.
Was die Progression trotz Therapie begünstigt
Generelle Risikofaktoren
- Usual-interstitial-pneumonia (UIP)-Muster der Fibrose
- ausgedehnte Traktionsbronchiektasen im HRCT
- rasche Krankheitsprogression
- keine Besserung oder Stabilisierung unter initialer Therapie
- Vorliegen eines short telomere syndroms
- höheres Alter
- bei systemischer Sklerose: höheres Alter zum Zeitpunkt der Diagnose, kurzer Krankheitsverlauf, schwarzamerikanische Ethnizität, gastroösophagealer Reflux
- bei rheumatoider Arthritis: Rauchen
- bei rheumatoider Arthritis und systemischer Sklerose: ausgedehnte interstitielle Lungenerkrankung im HRCT
- bei chronischer Hypersensitivitäts-Pneumonitis: kein identifizierbares Antigen, steigendes Alter
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).
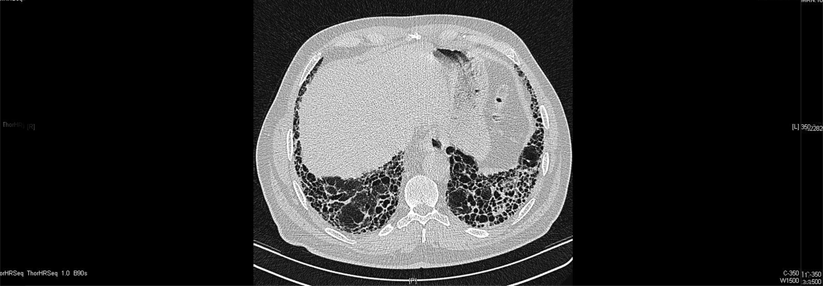



 Nicht nur bei der IPF kommt es zur progredienten Lungenfibrose.
© Science Photo Library/Alvin Telser
Nicht nur bei der IPF kommt es zur progredienten Lungenfibrose.
© Science Photo Library/Alvin Telser