Unterschiedliche Manifestationen, zahlreiche Therapiemöglichkeiten
 Die meisten MCL-Patienten benötigen früher oder später eine Salvage-Therapie. Bei symptomatischer lokalisierter Erkrankung kann eine Strahlentherapie erwogen werden.
© iStock/David A Litman
Die meisten MCL-Patienten benötigen früher oder später eine Salvage-Therapie. Bei symptomatischer lokalisierter Erkrankung kann eine Strahlentherapie erwogen werden.
© iStock/David A Litman
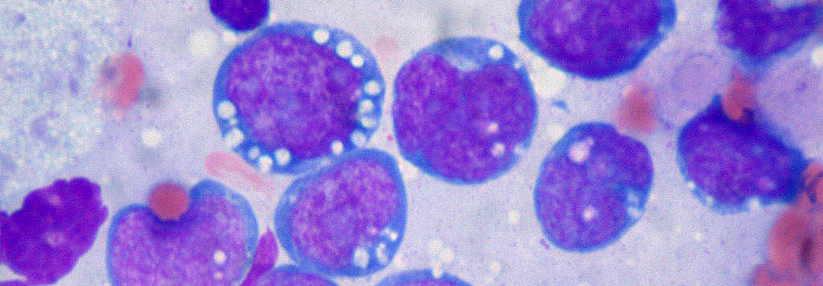
Das Mantelzelllymphom (MCL) macht etwa 5–7 % aller Lymphome aus. Das mediane Erkrankungsalter liegt zwischen 60 und 70 Jahren, wobei in etwa 70 % Männer von diesem Lymphomtyp betroffen sind, so Prof. Dr. James Armitage und Prof. Dr. Dan Longo von der University of Nebraska in Omaha. In vielen Fällen liegt die reziproke chromosomale Translokation t(11;14)(q13;32) vor, d.h. das BCL-1-Gen auf Chromosom 11 und das Gen für die schwere Immunglobulinkette auf Chromosom 14 haben die Plätze getauscht. Dies führt zu einer Überexpression des Proteins Cyclin-D1 und auf diese Weise zur Zellproliferation. Darüber hinaus sind weitere genetische Alterationen bei einem Teil der MCL-Betroffenen beschrieben.
Die meisten Patienten mit Mantelzelllymphom entwickeln eine palpable Lymphadenopathie mit oder ohne systemische Symptome, die oft lange Zeit unbeachtet bleibt. Zum Zeitpunkt der Diagnosestellung liegt deshalb bei einem Großteil der Betroffenen bereits eine weit fortgeschrittene Erkrankung vor, oftmals mit Knochenmarkbeteiligung. Etwa jeder dritte Patient berichtet über Fieber, Nachtschweiß oder Gewichtsverlust, noch häufiger wird über Fatigue geklagt. Bei ungefähr jedem vierten Patienten sind die Lymphknoten bis zu einem Durchmesser ≥ 10 cm stark vergrößert, weniger als die Hälfte der Betroffenen weisen erhöhte LDH-Spiegel auf. Eine ZNS-Beteiligung ist bei Erstmanifestation selten, geht aber mit einer sehr kurzen Überlebenszeit einher.
Manifestationen von Splenomegalie bis Polyposis
Das Mantelzelllymphom kann sich auch mit einer Splenomegalie, Knochenmarkbefall und zirkulierenden Lymphomzellen, jedoch ohne vergrößerte Lymphknoten, präsentieren. Diese Lymphome weisen oft einen (zunächst) indolenten klinischen Verlauf auf. Eine weitere ungewöhnliche Manifestation des Mantelzelllymphoms ist die lymphomatöse Polyposis des Gastrointestinaltrakts. Meist sind diese Polypen im distalen Ileum und Kolon lokalisiert.
Das Staging der Erkrankung orientiert sich an der Ann-Arbor- oder der Lugano-Klassifikation. Erfolgt im Rahmen der Diagnostik routinemäßig eine Positronenemissionstomografie, können mindestens 80 % der Patienten bereits in Stadium III oder IV eingestuft werden. Führt man zusätzlich Knochenmarkbiopsien oder Biopsien des Magen-Darm-Trakts durch, erhöht sich der Anteil mit Stadium IV. Am Ende der geplanten Therapie sollte ein Re-Staging erfolgen, bei dem man alle Tests wiederholt, die initial positiv waren. Zusätzlich steht eine Knochenmarkbiopsie an, falls noch nicht geschehen.
Wie wird das Mantelzelllymphom behandelt? Bei manchen Patienten genügt zunächst eine Watch-and-wait-Strategie. So sind Patienten mit Splenomegalie, Knochenmarksbeteiligung und zirkulierenden Lymphomzellen, aber ohne Lymphadenopathie, meist asymptomatisch und benötigen keine sofortige Therapie. Das trifft auch auf Patienten mit nodaler Manifestation und Low-Volume-Erkrankung, aber ohne Symptome, zu.
Für die Initialtherapie unterteilt man die Betroffenen häufig in zwei Gruppen:
- jüngere Patienten, die für eine konsolidierende autologe Knochenmarktransplantation fit genug sind
- ältere Patienten, die für eine Hochdosis-Chemotherapie und für eine autologe Transplantation eher nicht infrage kommen
Therapieziel ist für beide Gruppen eine komplette Remission. Transplantationskandidaten erhalten initial z.B. eine Therapie mit R-CHOP (Rituximab, Cyclophosphamid, Doxorubicin, Vincristin und Prednison) oder mit R-CHOP plus hoch dosiertem Cytarabin.
Rituximab spielt eine Schlüsselrolle
Daran schließt sich eine autologe Stammzelltransplantation mit oder ohne Rituximab-Erhaltungstherapie an. Patienten, die für eine Transplantation nicht geeignet sind, bekommen z.B. ebenfalls R-CHOP oder ein rituximabbasiertes Protokoll in Kombination mit Bendamustin, Lenalidomid oder Ibrutinib. Danach sollte eine Rituximab-Erhaltungstherapie in Betracht gezogen werden.
Die meisten MCL-Patienten benötigen früher oder später eine Salvage-Therapie. Bei symptomatischer lokalisierter Erkrankung kann eine Strahlentherapie erwogen werden. Für alle anderen ist eine systemische Zweitlinientherapie erforderlich, eventuell gefolgt von einer allogenen Transplantation. Patienten, die für eine Transplantation nicht geeignet sind, können z.B. eine Therapie mit neuen zielgerichteten Substanzen erhalten und – bei Therapieversagen – eine Behandlung mit CAR-T-Zellen.
Quelle: Armitage JO, Longo DL. N Engl J Med 2022; 386: 2495-2506; DOI: 10.1056/NEJMra2202672
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).



 Die meisten MCL-Patienten benötigen früher oder später eine Salvage-Therapie. Bei symptomatischer lokalisierter Erkrankung kann eine Strahlentherapie erwogen werden.
© iStock/David A Litman
Die meisten MCL-Patienten benötigen früher oder später eine Salvage-Therapie. Bei symptomatischer lokalisierter Erkrankung kann eine Strahlentherapie erwogen werden.
© iStock/David A Litman