Mukoviszidose: Neue Ansätze in der Behandlung
 Zwar bleibt Mukoviszidose die häufigste letale autosomal-rezessive Erbkrankheit, aber es gibt Fortschritte.
© iStock/Steve Debenport
Zwar bleibt Mukoviszidose die häufigste letale autosomal-rezessive Erbkrankheit, aber es gibt Fortschritte.
© iStock/Steve Debenport
Die Mukoviszidose (zystische Fibrose, CF) betrifft etwa eins von 4000 Neugeborenen. Zwar bleibt sie die häufigste letale autosomal-rezessive Erbkrankheit, aber es gibt Fortschritte: Das mittlere Sterbealter hat sich von rund 5 Jahren in den 1960er-Jahren auf derzeit etwa 35 Jahre erhöht. Heute geborene Kinder mit Mukoviszidose werden wahrscheinlich im Durchschnitt ein Alter von über 50 Jahren erreichen. Seit 2016 gehört die CF zum Neugeborenenscreening in Deutschland.
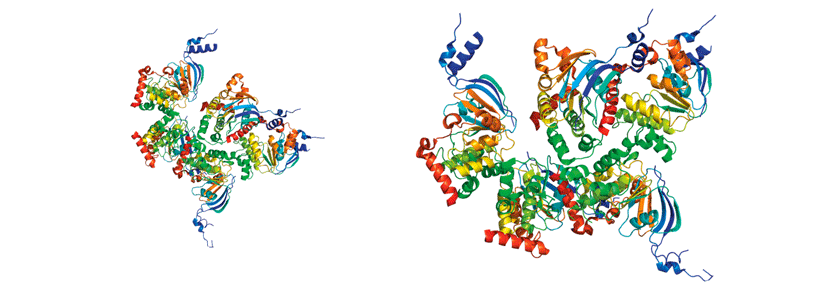
Die Mukoviszidose beruht auf Mutationen im CFTR(cystic fibrosis transmembrane regulator)-Gen auf Chromosom 7. Das CFTR-Protein wird in allen exokrinen Drüsen exprimiert und fungiert als Anionenkanal für Chlorid und…
Liebe Leserin, lieber Leser, aus rechtlichen Gründen ist der Beitrag, den Sie aufrufen möchten, nur für medizinische Fachkreise zugänglich. Wenn Sie diesen Fachkreisen angehören (Ärzte, Apotheker, Medizinstudenten, medizinisches Fachpersonal, Mitarbeiter der pharmazeutischen oder medizintechnischen Industrie, Fachjournalisten), loggen Sie sich bitte ein oder registrieren sich auf unserer Seite. Der Zugang ist kostenlos.
Benutzeranmeldung
Bitte geben Sie Ihren Benutzernamen und Ihr Passwort ein, um sich an der Website anzumelden.
Bei Fragen zur Anmeldung senden Sie bitte eine Mail an online@medical-tribune.de.