Amyloid-Kardiomyopathie früh behandeln
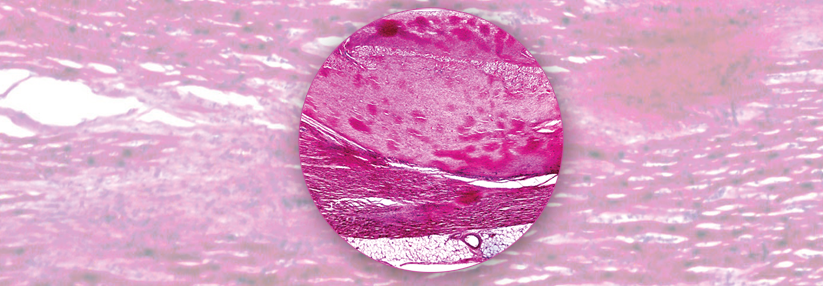 Lichtmikroskopisch sind in der Herzmuskulatur dunkel-pink gefärbte Amyloidablagerungen zu erkennen.
© Science Photo Library/Biophoto Associates
Lichtmikroskopisch sind in der Herzmuskulatur dunkel-pink gefärbte Amyloidablagerungen zu erkennen.
© Science Photo Library/Biophoto Associates
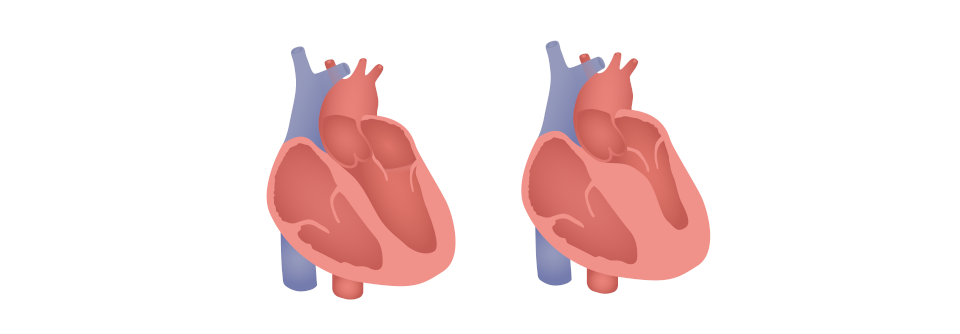
Bei der Amyloidose des Herzens entwickelt sich eine restriktive Kardiomyopathie, weil Proteine extrazellulär im Myokard abgelagert werden. Meist handelt es sich um fehlgefaltete Immunglobulin-Leichtketten (AL) oder um pathologische Varianten von Transthyretin (TTR). Bei Letzterem handelt es sich um ein in der Leber synthetisiertes Protein, das normalerweise beim Transport von Thyroxin und retinolbindendem Protein eine Rolle spielt.
TTR-Amyloid lagert sich in vielen Organen ab
In der Praxis ist vor allem die Transthyretin-Amyloid-Protein-Kardiomyopathie (ATTR-CM) bedeutsam, heißt es in einem Scientific Statement der American Heart Association unter Federführung von Dr. Michelle M. Kittleson vom Cedars-Sinai California Heart Center in Beverly Hills. Sie kann auf dem Boden von autosomal dominant vererbten Veränderungen des Transthyretin-Gens entstehen. Häufiger ist jedoch die Deposition von Wildtyp-Transthyretin-Protein, was man früher als senile kardiale Amyloidose bezeichnet hat.
Das TTR-Amyloid lagert sich auch in anderen Organen ab. So beobachtet man bei betroffenen Patienten häufig ein Karpaltunnelsyndrom, eine lumbale Spinalstenose, Bizepssehnenrupturen und autonome oder sensorische Polyneuropathien.
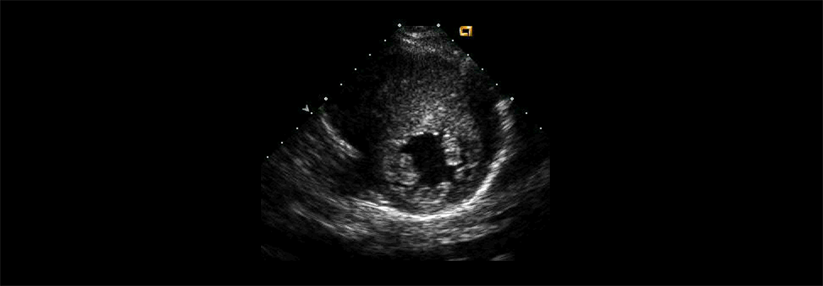
Die Kranken präsentieren sich meist mit Dyspnoe, Fatigue und Ödemen. Häufig wird dann fälschlicherweise die Diagnose Herzinsuffizienz mit erhaltener Auswurffraktion (HFpEF) gestellt. Zeigt das Echokardiogramm eine erhebliche Wandverdickung des linken Ventrikels (≥ 14 mm), wird oft eine hypertensive Kardiomyopathie angenommen. Doch man sollte eben auch an andere Ursachen wie die Amyloidose denken. Auf sie kann eine Diskrepanz zwischen erheblicher Wandverdickung und niedriger QRS-Voltage im EKG hinweisen.
Weitere Verdachtsmomente liefern u.U. eine positive Familienanamnese bzgl. Kardiomyopathie, eine persistente niederschwellige Troponin-Erhöhung sowie eine Intoleranz gegenüber Antihypertensiva und Herzinsuffizienzmedikamenten, die sich mit Hypotension oder Orthostase manifestiert.
Echokardiographie und Magnetresonanztomographie können den Verdacht auf eine kardiale Amyloidose weiter untermauern. Mit der Technetium-Szintigraphie gelingt es heute, die Diagnose der ATTR-CM zu sichern – in der Regel ohne bestätigende Endomyokard-Biopsie. Die Technetium-Tracer binden auf bisher ungeklärte Weise an das Amyloid. Das tun sie allerdings auch bei der AL-Amyloidose. Diese muss deshalb durch ein Screening auf freie Leichtketten in Serum und Urin ausgeschlossen werden.
Der nächste Schritt in der Diagnostik ist die Sequenzierung des TTR-Gens. Es muss geklärt werden, ob es sich um mutiertes oder Wildtyp-ATTR-Protein handelt. Denn nur wenn Mutationen vorliegen, ist eine genetische Beratung notwendig. Zudem sind manche Therapien nur in sochen Fällen zugelassen.
Nachdem die Mechanismen der Amyloidbildung besser aufgeklärt werden konnten, hat sich in den letzten Jahren in der Therapie etwas getan. Entwickelt und zugelassen wurden Substanzen, die an der TTR-Produktion (Silencer), der Dissoziation (Stabilisatoren) und der Clearance aus dem Gewebe (Disruptoren) ansetzen. Silencer wie Patisiran (siRNA) und Inotersen (Antisense-Nukleotid) hemmen die hepatische Synthese des pathologischen Proteins. Beide verringern die Konzentration von zirkulierendem TTR um mehr als 85 %. Sie verlangsamen auch die Progression der Amyloidose-assoziierten Polyneuropathie. Weiter gibt es Hinweise auf positive kardiale Effekte wie eine Abnahme der linksventrikulären Wanddicke.
Therapie in der Praxis
Bei vorwiegend kardialer ATTR- Amyloidose (mutiert oder Wildtyp) und einer Herzinsuffizienz im Stadium NYHA I-III ist Tafamidis indiziert.
Patisiran oder Inotersen kommen in Betracht für Patienten mit einer Amyloidose durch mutiertes ATTR-Protein und Polyneuropathie mit oder ohne Kardiomyopathie. Keine der beiden Substanzen eignet sich für die ATTR-Kardiomyopathie vom Wildtyp.
Diflunisal kann off label eingesetzt werden bei asymptomatischen ATTR-Trägern oder bei Patienten mit ATTR-Kardiomyopathie bzw. -Neuropathie, die andere Therapien aus verschiedenen Gründen nicht bekommen können.
Gesamtmortalität kann deutlich reduziert werden
TTR-Stabilisatoren binden an das TTR-Tetramer und verhindern die Fehlfaltung des Proteins und damit die Deposition von Amyloidfibrillen. Zu ihnen gehören Diflunisal und Tafamidis. Während für Diflunisal nur eine verlangsamte Progression der Polyneuropathie belegt ist, konnte Tafamidis in einer randomisierten Studie die Gesamtmortalität der Patienten mit ATTR-Kardiomyopathie und die Rate von Krankenhausaufnahmen aus kardialer Ursache signifikant senken. Auch die 6-Minuten-Gehstrecke und der Kansas-Kardiomyopathie-Score nahmen unter dieser Therapie signifikant langsamer ab. Disruptoren konnten sich wegen einer hohen Rate an Nebenwirkungen und fraglicher Effektivität bisher nicht etablieren. Es liegt auf der Hand, dass sich mit diesen spezifischen Therapien am meisten erreichen lässt, wenn damit begonnen wird, bevor die kardiale Dysfunktion signifikant geworden ist. Deshalb kommt es entscheidend darauf an, Betroffene frühzeitig zu erkennen. Basis der Therapie der ATTR-Kardiomyopathie muss über die krankheitsmodifizierenden Medikamente hinaus immer ein gutes Management von Herzinsuffizienz und Arrhythmien sein.Quelle: Kittleson MM et al. Circulation 2020; DOI: 10.1161/CIR.0000000000000792
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).



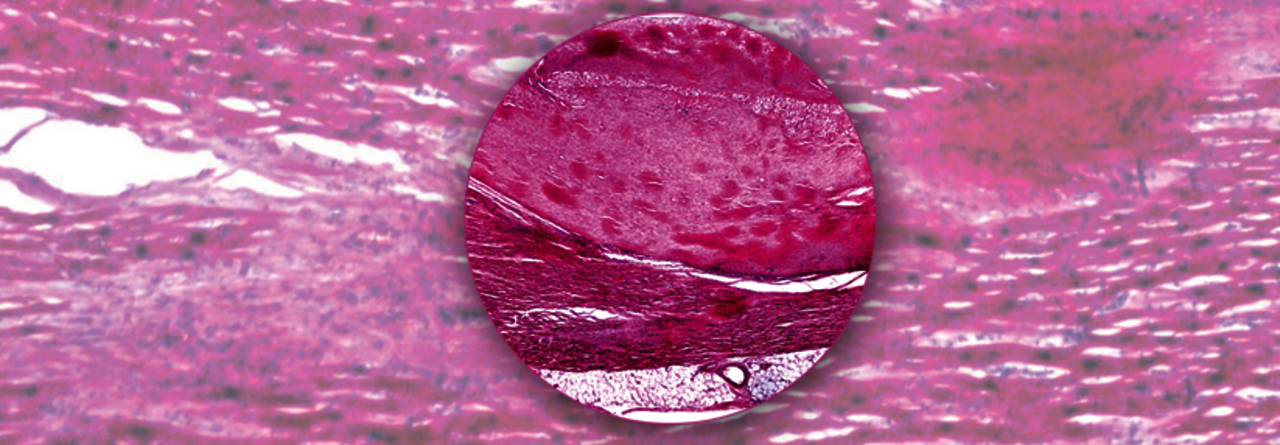 Lichtmikroskopisch sind in der Herzmuskulatur dunkel-pink gefärbte Amyloidablagerungen zu erkennen.
© Science Photo Library/Biophoto Associates
Lichtmikroskopisch sind in der Herzmuskulatur dunkel-pink gefärbte Amyloidablagerungen zu erkennen.
© Science Photo Library/Biophoto Associates