Transthyretin-Amyloidose: Neue Therapieoptionen im Überblick
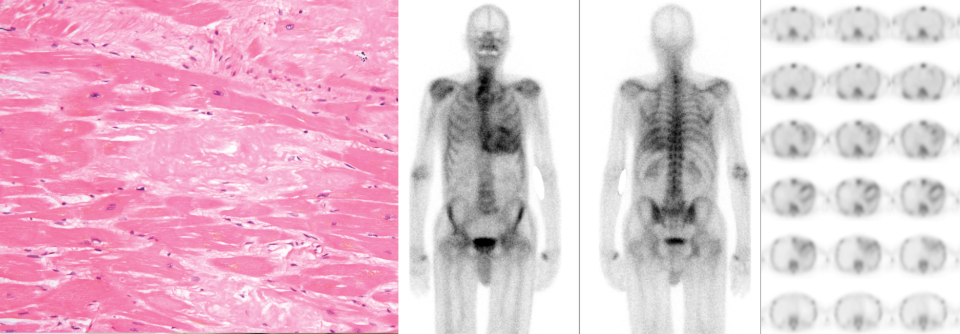
 In Deutschland hat jeder Zweite mit ATTR-Amyloidose eine Kardiomyopathie.
© iStock/MicroStockHub
In Deutschland hat jeder Zweite mit ATTR-Amyloidose eine Kardiomyopathie.
© iStock/MicroStockHub
Grob werden zwei Typen der Transthyretin(TTR)-assoziierten Amyloidose unterschieden, erklärte Professor Dr. Claudio Rapezzi, Universität Bologna. Die hereditäre Form basiert auf angeborenen Mutationen im Transthyretin-Gen und kann nahezu alle Gewebe infiltrieren, manifestiert sich aber insbesonders an Nervensystem und Herz. Die Wildtyp-Form ist homogener, sie befällt praktisch immer das Myokard.
Die Befallsmuster divergieren erheblich – sogar innerhalb Europas: Während in Deutschland jeder zweite Patient mit ATTR-Amyloidose an einer Kardiomyopathie leidet und nur ein Drittel an isolierten neurologischen Symptomen, machen Letztere z.B. in Portugal 80 % der Fälle aus. Ein gemischter Phänotyp zeigt sich in diesen beiden Ländern bei 20 % bzw. 16 % der Betroffenen.
Mehr als 90 % des Transthyretins entstehen in der Leber in Form von Tetrameren, die anschließend in Monomere zerlegt werden. Neben der Transthyretinsynthese limitiert die Dissoziationsrate das Ausmaß, in dem fehlgefaltete Monomere zu Amyloidfibrillen aggregieren und sich in den Geweben ablagern. Die jetzt verfügbaren Wirkstoffe setzen an beiden Schritten an.
Die Präparate legen die für die TTR-Synthese zuständigen mutierten Gene durch RNA-Interferenz (Patisiran) oder Antisense-Oligonukleotide (Inotersen) still bzw. stabilisieren das Protein selektiv auf der Tetramerstufe (Tafamidis). Die auf Genebene wirkenden Substanzen eignen sich für die hereditäre ATTR-Amyloidose, der Stabilisator setzt weiter unten in der Kaskade an und ist daher auch beim Wildtyp wirksam.
Alle drei Wirkstoffe haben ihre Effektivität in randomisierten klinischen Doppelblindstudien unter Beweis gestellt, die letztes Jahr veröffentlicht wurden. Die von Prof. Rapezzi geleitete ATTR-ACT-Studie mit Tafamidis konnte bei ATTR-Kardiomyopathie sogar eine Senkung der Mortalität** nachweisen, die sich bereits nach 18 Monaten manifestierte. Der Kollege betonte, dass die Therapie möglichst früh beginnen sollte, „sonst verbringt der Patient die gewonnene Lebenszeit im Krankenhaus“.
Auch APOLLO (Patisiran) und Neuro-TTR (Inotersen) zeigten bei hereditärer Transthyretin-Amyloidose eindrucksvoll, was sich mit den neuen Therapien erreichen lässt. Jeweils über die Hälfte der Teilnehmer hatten kardiale Auffälligkeiten. Mortalität war hier kein Endpunkt, aber in beiden Studien kam u.a. der Progress der neurologischen Symptome praktisch zum Stillstand. Wichtig: Durch keine der genannten Studien kamen Sicherheitsbedenken auf.
Wachsende Therapieoptionen
Wildtyp-ATTR-Kardiomyopathie:
- Tafamidis
- ausschließlich Kardiomyopathie: Tafamidis
- Kardiomyopathie UND Neuropathie: Tafamidis, Patisiran, Inotersen
- ausschließlich Neuropathie: Patisiran, Inotersen
Modifiziert nach: Ruberg FJ et al. J Am Coll Cardiol 2019; 73: 2872-2891
Wirkstoffe (noch) nicht für alle Ausprägungen zugelassen
US-Experten haben bereits eine Übersicht für die Behandlung der ATTR-Amyloidosen erarbeitet (s. Kasten). Sie setzt allerdings voraus, dass die Wirkstoffe für alle Ausprägungen der Erkrankung zugelassen sind, was in Deutschland (noch) nicht der Fall ist. Hierzulande dürfen sie on label derzeit nur bei ATTR-Amyloidose mit Polyneuropathie eingesetzt werden, Patisiran und Inotersen nur bei der hereditären Form.Quelle: ESC* Congress 2019
* European Society of Cardiology
** Herztransplantation und Implantation eines Herzunterstützungssystems wurden in dieser Analyse als Tod gewertet
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).




 In Deutschland hat jeder Zweite mit ATTR-Amyloidose eine Kardiomyopathie.
© iStock/MicroStockHub
In Deutschland hat jeder Zweite mit ATTR-Amyloidose eine Kardiomyopathie.
© iStock/MicroStockHub