Non-Hodgkin-Lymphom - Zielgerichtete Wirkstoffe und neue Kombinationstherapien bessern die Prognose
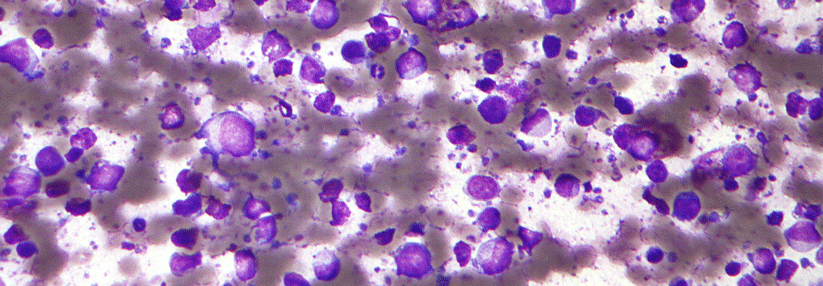
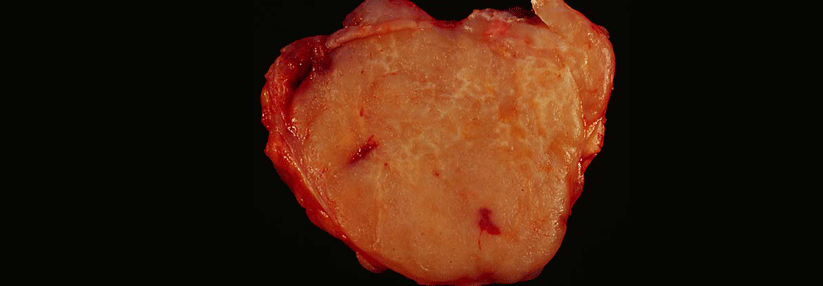
 Burkitt-Lymphom.
© wikimedia/Mike Blyth
Burkitt-Lymphom.
© wikimedia/Mike Blyth
Grundsätzlich lassen sich Non-Hodgkin-Lymphome (NHL) in indolente (niedrigmaligne) und aggressive (hochmaligne) Formen unterteilen. Sie stammen von Lymphozyten in den unterschiedlichsten Entwicklungsstadien ab – etwa 85 bis 90 % von B-Zellen, der Rest von T-Zellen oder natürlichen Killerzellen, schreibt Professor Dr. James O. Armitage aus Omaha.
Zahlreiche Faktoren stehen mit einem erhöhten Risiko für NHL in Zusammenhang. Dazu gehören u.a. Autoimmunerkrankungen und bestimmte virale und bakterielle Krankheiten. So erhöht eine Infektion mit H. pylori das Risiko für MALT-Lymphome, das Epstein-Barr-Virus die Wahrscheinlichkeit für ein Burkitt-Lymphom und Hepatitis C die für ein diffuses…
Liebe Leserin, lieber Leser, aus rechtlichen Gründen ist der Beitrag, den Sie aufrufen möchten, nur für medizinische Fachkreise zugänglich. Wenn Sie diesen Fachkreisen angehören (Ärzte, Apotheker, Medizinstudenten, medizinisches Fachpersonal, Mitarbeiter der pharmazeutischen oder medizintechnischen Industrie, Fachjournalisten), loggen Sie sich bitte ein oder registrieren sich auf unserer Seite. Der Zugang ist kostenlos.
Benutzeranmeldung
Bitte geben Sie Ihren Benutzernamen und Ihr Passwort ein, um sich an der Website anzumelden.
Bei Fragen zur Anmeldung senden Sie bitte eine Mail an online@medical-tribune.de.